
Abstract
Background
Congenital hemidysplasia with ichthyosiform erythroderma and limb defects also known as CHILD syndrome is an X-linked dominant, male lethal genodermatosis with a prevalence of 1 in 100,000 live births. Mutations in NSDHL gene located at Xq28 potentially impair the function of NAD(P) H steroid dehydrogenase-like protein and is responsible for its pathogenesis.
Case presentation
The proband was a 9-month-old twin (T2) girl with a healthy twin sister (T1) of Sri Lankan origin born to non-consanguineous parents. She presented with right sided continuous icthyosiform erythroderma and ipsilateral limb defects and congenital hemidysplasia since birth. Notably the child had ipsilateral hand hypoplasia and syndactyly. There were other visceral abnormalities. We performed whole exome sequencing and found a novel heterozygous variant (NSDHL, c.713C > A, p.Thr238Asn).
Conclusion
We report a novel missense variant in the NSDHL gene that resides in a highly-conserved region. This variant affects the NAD(P) H steroid dehydrogenase-like protein function via reduction in the number of active sites resulting in the CHILD syndrome phenotype and syndactyly.
Similar content being viewed by others


Background
Congenital hemidysplasia with ichthyosiform erythroderma and limb defects also known as CHILD syndrome is an X-linked dominant, male lethal genodermatosis with a prevalence of 1 in 100,000 live births. It is characterized by unilateral inflammatory skin lesions, ipsilateral limb and visceral abnormalities [1, 2]. Thus, so far only 60 cases are reported in literature with twice as more right side involvement than the left. The hallmark skin lesions in this condition is psoriasiform epidermis with hyperkeratosis and parakeratosis along with foam cell infiltration of the papillary dermis. These skin abnormalities are present at birth and are persistent throughout life. However, lesions seem to spare the face and are more commonly seen between the skin folds and creases. Visceral abnormalities may involve the cardiovascular, respiratory, nervous and renal systems. Mutations in NSDHL gene located at Xq28 potentially impair the function of NAD(P) H steroid dehydrogenase-like protein which is responsible for its pathogenesis. It is an essential enzyme in the cholesterol biosynthesis pathway [3, 4]. NAD(P) H steroid dehydrogenase-like protein is a C4 demethylase and catalyzes NAD+ dependent oxidative decarboxylation of the C4 methyl groups of 4α-carboxysterols [3, 4]. The latter is involved in post-squalene cholesterol biosynthesis and is also a regulator of the Epidermal Growth Factor Receptor (EGFR) trafficking pathways. There is a dual localization of this protein, within the membranes of endoplasmic reticulum and on the surface of lipid droplets. Dysregulation of this pathway has also shown to cause metabolic derangements such as hypercholesterolemia, certain cancers, cardiovascular diseases and neuropathies [5,6,7]. Here we report the first Sri Lankan patient with CHILD syndrome due to a novel heterozygous variant (NSDHL, c.713C > A, p.Thr238Asn).
Case presentation
The proband was a 9-month-old twin (T2) girl of Sri Lankan origin born to non-consanguineous parents and her twin sister (T1) was healthy. She presented with congenital hemidysplasia of the right side, ipsilateral limb defects and icthyosiform erythroderma which was continuous from her neck to mid-thigh including the upper arm. There were other visceral abnormalities such as; an absent right (ipsilateral) kidney and.
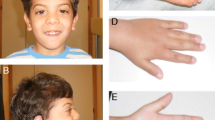
asymmetrical ventriculomegaly (L > R) in the brain scan. However, we noticed that the child had hypoplastic hands with syndactyly a feature which was not reported in literature previously (Table 1). We performed whole exome sequencing and found a novel heterozygous variant (NSDHL, c.713C > A, p.Thr238Asn). She was diagnosed with Congenital hemidysplasia with ichthyosiform erythroderma and limb defects (CHILD) syndrome, in-silico analysis predicted this as a pathogenic variant. She had undergone traditional Sri Lankan herbal medicine in the form of topical and oral preparations for the skin lesions for less than a year. Which has shown significant improvement (Fig. 1a,b,c). However, the exact details of the preparations used was not disclosed.

a–Proband at 9 months with ipsilateral ichthyosiform erythroderma and Hypoplasia of arm and hand. b- Proband after 1 year, following treatment. Images showing remarkable improvement in the skin lesions. c- Simple syndactyly of right hand with nail changes
In-silico analysis
The initial analysis was done using the National Center for Biotechnology Information (NCBI) database, mRNA sequence of NAD(P) H steroid dehydrogenase-like protein (NSDHL)(XM_011531178.2; https://www.ncbi.nlm.nih.gov/nuccore/XM_011531178.2) was retrieved in FASTA format from NCBI with NCBI Reference Sequence ID: NP_057006.1 having 373 amino acids. Observed variation in proband was done manually in the mRNA sequence and taken for further analysis as c.713C > A, p.Thr238Asn.
UniProt database for protein sequences and functional information revealed the Protein sequence of NAD(P) H steroid dehydrogenase-like protein (NSDHL) which was retrieved in FASTA format from UniProtKB repository having unique ID Q15738 (NSDHL_HUMAN). This protein sequence was taken as a reference sequence for comparisons of variant protein due to variations in DNA sequence and in protein sequence. There were also some previously reported variations in the Uniprot database and they were p.Alar105Val, p.Ala182Pro, p.Gly205Ser [10]. I-Tasser an online bioinformatics server for the prediction of protein structure and function (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) was then used to form the 3D construct [11]. I-TASSER detects templates from PDB and constructs full length protein models by replica exchange Monte-Carlo simulations. Sequences of normal and mutated NSDHL protein were submitted to I-TASSER for 3D structure prediction and predicted protein models were downloaded and analyzed. Followed by automated homology modeling using the Swiss-PdbViewer [12]. Both the proteins were then compared with reference to active sites present in them. Stereochemical quality of the protein structure was then analyzed using PROCHECK. Thus, a Ramachandran plot was obtained which showed energetically allowed regions for backbone dihedral angles ψ against φ of amino acid residues in protein structure [11].
In silico analysis using nine computational prediction tools namely DANN, DEOGEN2, FATHMM-MKL, M-CAP, MVP, MutationAssessor, MutationTaster, REVEL and SIFT [13, 14] returned results of likely pathogenic effects due to NSDHL, c.713C > A, p.Thr238Asn This variant is also not found in all known population databases or in our in-house Sri Lankan population database (Supplementary file).
Further, after modeling normal and the NSDHL proteins of the proband we found that there is a difference in the secondary and tertiary structure. Number of alpha helices and beta sheets were different in both proteins. There is a difference in the starting and the ending positions of the secondary structures and also in the spatial arrangement of alpha helices and beta sheets in the 3D folding. The accuracy of secondary structures was also perfect and supported by value of normalized B-factor. Normalized B-factor for a target protein is defined as z-score-based normalization of the raw B-factor values. In our predicted model majority of secondary structures had less than 0 values indicating its stability. The estimated normalized B-factor is shown in Fig. 2a & b. We compared the Ramachandran plots of the wild-type protein and mutated protein (Fig. 3a & b). The wild type protein had residues in the following region of the plot, 250 (73.7%) in the preferred regions such as A, B, L; 61 (18.0%) in a,b,l,p regions; 24 (7.1%) in regions ~a,~b,~l,~p and only 4 (1.2%) in unfavorable regions. In the mutant proteins we observed 242 (71.4%) in A, B, L; 76 (22.4%) in a,b,l,p regions; 16 (4.7%) in regions ~a,~b,~l,~p and 5 (1.5%)) in unfavorable regions. We also observed that the variant found in the proband reduced the number of active sites in the resultant protein. Normal protein has 9 active sites whereas variant protein has only 6 active sites (Fig. 4a & b). We also analyzed the differences in the protein surface to determine the type of interactions. The difference in the cavities between the wild type and the mutant protein is described in Table 2A and B respectively. As per our analysis we found less number of cavities/ pockets in the mutated protein. Thus, reducing its ligand binding potential. The exact pathways that these ligands facilitate is yet to be determined. However, this leads to a reduction in the number of interacting molecules. Hence, we speculate that there is a significant reduction in the protein function.

a- Distribution and B factor values of alpha helices and beta sheets in wild type protein,b - In mutant protein both show similar distribution patterns

a- Ramachandran plot of wild type NHDL protein; The wild type protein have residues in the following region of the plot, 250 (73.7%) in the preferred regions such as A, B, L; 61 (18.0%) in a,b,l,p regions; 24 (7.1%) in regions ~a,~b,~l,~p and only 4 (1.2%) in unfavorable regions. b- Ramachandran plot of mutant NHDL protein; In the mutant proteins we observed 242 (71.4%) in A, B, L; 76 (22.4%) in a,b,l,p regions; 16 (4.7%) in regions ~a,~b,~l,~p and 5 (1.5%)) in unfavorable regions

a: Wild type NSDHL protein structure with nine active sites, b Mutant NSDHL protein structure with only six active sites
Discussion & conclusion
CHILD syndrome is a X linked dominant condition with multisystem involvement showing a unique lateralization pattern and a strict midline demarcation of an inflammatory epidermal nevus. NAD(P) H steroid dehydrogenase-like protein (Nsdhl), which is responsible for this condition is a C3 sterol dehydrogenase enzyme involved in the conversion of lanosterol into cholesterol. Hence, the CHILD phenotype could result from the lack of cholesterol or other sterols downstream due to the block in its biosynthesis or due to the accumulation of products upstream to this pathway. In cell lines where a missense variant in the NSDHL gene was introduced, it was found that there was a significant interruption to this pathway as compared to wild-type NSDHL cell lines [8, 15]. In keeping with the current literature, molecular genetic analysis in our patient strongly predicted that CHILD syndrome was caused by a novel missense variant in NSDHL it was also found that the variant was located in a highly-conserved site in the NAD(P) H steroid dehydrogenase-like protein across other species as well (Table 3). Syndactyly was a unique feature we observed in this child and it could be due to cholesterol metabolism pathways acting downstream of cytochrome P450 oxidoreductase enzyme and its involvement in skeletal development [16].
By analyzing the Ramachandran plot of the two proteins we observed that its structural stability of the protein was unaffected. However, through in-silico protein modeling studies we also observed that the wild type protein had 9 active sites whereas the mutated protein in our patient had only 6. Hence, we suspect the 3 missing active sites are crucial for its enzymatic function. All computational and predictive analyses thus indicate NSDHL, c.713C > A, p.Thr238Asn as a null variant resulting in loss of function of NSDHL gene that is already known to lead to CHILD syndrome. This report contributes to the growing list of missense variants that have been associated with CHILD syndrome [8, 9] and facilitates discovery of more patients with similar variant.
We report a novel missense variant in the NSDHL gene residing in a highly-conserved region which affects the NAD(P) H steroid dehydrogenase-like protein function via reduction in the number of active sites. This variant expands the phenotype and genotype data suspected to be associated with CHILD syndrome.
Availability of data and materials
The datasets generated and analysed during the current study are available in GenBank accession number: MT747166, link- https://www.ncbi.nlm.nih.gov/nuccore/MT747166
Abbreviations
- SPDBV:
-
Swiss-PdbViewer
- I-TASSER:
-
Iterative Threading ASSEmbly Refinement
- NCBI:
-
National Center for Biotechnology Information
- EGFR:
-
Epidermal Growth Factor Receptor
References
König A, et al. Mutations in the NSDHL gene, encoding a 3β-hydroxysteroid dehydrogenase, cause CHILD syndrome. Am J Med Genet. 2000;90(4):339–46..
"Orphanet: Congenital Hemidysplasia With Ichthyosiform Erythroderma And Limbs Defects". Orpha. Net, 2020, https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=2136&Disease_Disease_Search_diseaseType=ORPHA&Disease_Disease_Search_diseaseGroup=139&Disease(s)/group%20of%20diseases=Congenital-hemidysplasia-with-ichthyosiform-erythroderma-and-limbs-defects&title=Congenital-hemidysplasia-with-ichthyosiform-erythroderma-and-limbs-defects&search=Disease_Search_Simple. Accessed 8 Apr 2020.
Mi X-b, et al. CHILD syndrome: case report of a Chinese patient and literature review of the NAD [P] H steroid dehydrogenase-like protein gene mutation. Pediatr Dermatol. 2015;32(6):e277–82.
Hummel M, et al. Left-sided CHILD syndrome caused by a nonsense mutation in the NSDHL gene. Am J Med Genet A. 2003;122(3):246–51.
Caldas H, Herman GE. NSDHL, an enzyme involved in cholesterol biosynthesis, traffics through the Golgi and accumulates on ER membranes and on the surface of lipid droplets. Hum Mol Genet. 2003;12(22):2981–91.
Kim D-G, et al. Crystal structures of human NSDHL and development of its novel inhibitor with the potential to suppress EGFR activity. Cell Mol Life Sci. 2020:1–19.
Mo C, et al. Protein–protein interactions among C-4 demethylation enzymes involved in yeast sterol biosynthesis. Proc Natl Acad Sci. 2002;99(15):9739–44.
Bornholdt D, et al. Mutational spectrum of NSDHL in CHILD syndrome. J Med Genet. 2005;42(2):e17.
König A, et al. A novel missense mutation of NSDHL in an unusual case of CHILD syndrome showing bilateral, almost symmetric involvement. J Am Acad Dermatol. 2002;46(4):594–6.
Guex, Nicolas, and Manuel C. Peitsch. "SWISS-MODEL and the Swiss-Pdb Viewer: an environment for comparative protein modeling." Electrophoresis (1997) 18.15: 2714–2723.
Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER suite: protein structure and function prediction. Nat Methods. 2014;12(1):7–8. https://doi.org/10.1038/nmeth.3213.
Guex N, Peitsch MC, Schwede T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis. 2009;30(SUPPL. 1):162–73. https://doi.org/10.1002/elps.200900140.
Ohashi M, et al. Localization of mammalian NAD (P) H steroid dehydrogenase-like protein on lipid droplets. J Biol Chem. 2003;278(38):36819–29.
Bateman A. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47(D1):D506–15. https://doi.org/10.1093/nar/gky1049.
Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–91.
Schmidt K, et al. Cholesterol metabolism: the main pathway acting downstream of cytochrome P450 oxidoreductase in skeletal development of the limb. Mol Cell Biol. 2009;29(10):2716–29.
Acknowledgements
We wish to acknowledge the laboratory staff for their contribution.
Funding
No funding for this project.
Author information
Authors and Affiliations
Contributions
DH and VHWD are the clinicians looking after the patient. VHWD and PSL critically evaluated and guided the project. DH and PSL performed the bioinformatics analysis of the variant. HP contributed with in-silico protein modeling and bioinformatics approaches. DH and VHWD wrote the first draft of the manuscript with contributions from all. All authors reviewed modified and approved the final manuscript.
Authors’ information
Dr. Dineshani Hettiarachchi MBBS, MSc, MCGP, SEDA UK.
Is a lecturer at Department of Anatomy and Human Genetics Unit, Faculty of Medicine, University of Colombo, Sri Lanka.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics approval for the study was obtained by the Faculty of Medicine, University of Colombo, Ethics Review Committee and written informed consent for genetic testing was obtained from the parents the children as they were minors.
Consent for publication
Written consent for publication was obtained from the parents.
Competing interests
Authors declare that there are no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table 1.
Pathogenicity prediction and conservation scores for NSDHL c.713C>A p.Thr238Asn. Table 2. Frequency of NSDHL c.713C>A p.Thr238Asn in global population databases
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hettiarachchi, D., Panchal, H., Lai, P.S. et al. Novel variant in NSDHL gene associated with CHILD syndrome and syndactyly- a case report. BMC Med Genet 21, 164 (2020). https://doi.org/10.1186/s12881-020-01094-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-020-01094-y