
Abstract
Background
Wolf-Hirschhorn syndrome (WHS) is a contiguous gene syndrome caused by partial 4p deletion highly variable in size in individual patients. The core WHS phenotype is defined by the association of growth delay, typical facial characteristics, intellectual disability and seizures. The WHS critical region (WHSCR) has been narrowed down and NSD2 falls within this 200 kb region. Only four patients with NSD2 variants have been documented with phenotypic features in detail.
Case presentation
Herein, we report the case of a 12-year-old boy with developmental delay. He had dysmorphic facial features including wide-spaced eyes, prominent nasal bridge continuing to forehead, abnormal teething and micrognathia. He also had mild clinodactyly of both hands. Using whole-exome sequencing, we identified a pathogenic mutation in NSD2 [c.4029_4030insAA, p.Glu1344Lysfs*49] isolated from peripheral blood DNA. Sanger confirmation of this variant revealed it as a de novo truncating variant in the family.
Conclusion
Here, we reported a boy with de novo truncating variant in NSD2 with atypical clinical features comparing with 4p16.3 deletion related WHS. Our finding further supported the pathogenesis of truncating variants in NSD2 and delineated the possible symptom spectrum caused by these variants.
Similar content being viewed by others

Background
Wolf-Hirschhorn syndrome (WHS) was first described in 1965 as a congenital anomalies/mental retardation due to partial deletion on p-terminal of chromosome 4 [1]. WHS (OMIM 194190) patients were characterized with craniofacial features including microcephaly, ‘Greek warrior helmet’ appearance of wide nasal bridge, widely spaced and prominent eyes, and distinct mouth with down-turned corners, short philtrum and micrognathia [2]. Other frequent features observed in WHS patients were intrauterine growth retardation, postnatal growth deficiency, intellectual disability, hypotonia, seizures, feeding difficulties and muscle hypotrophy [3]. Depending mostly on the extent of the 4p deletion, additional clinical signs include major malformations, as midline defects, congenital heart defects, renal and skeletal anomalies [4].
Overlapping regions of multiple cases diagnosed with WHS has helped to decide the critical region of WHS, namely WHSCR1 and WHSCR2, which has been narrowed down to a 200 kb region on 4p16.3 [5, 6]. Typical WHS, even in the mild form of its clinical phenotype, is largely assumed to be a multigenic disorder. Thus, neither WHSCR nor WHSCR-2 was established as definite genetic cause of WHS, but they allowed further exploration of possible candidate genes. De novo variation in NSD2 (also known as WHSC1) is thought to be related to diseases since identified in patients with a wide range of phenotypic features including developmental delay, autism, and congenital cardiac disorders. Recent reported cases with de novo truncating variants on NSD2 and detail phenotypic features have offered a new insight into genetic causes of WHS [7,8,9]. NSD2 is thought to be the major, but not the unique gene for facial dysmorphisms, growth delay and intellectual disability. Here we report a boy with de novo variant in NSD2 with atypical WHS clinical manifestations, further supported the pathogenesis of truncating variant in NSD2 and delineated the possible symptom spectrum caused by these variants on a single gene.
Case presentation
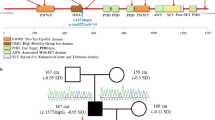
The patient, a 12-year-old boy, the only child of healthy unrelated parents, was referred to the department of developmental behavior of Shanghai Children’s Medical Center due to delayed development and growth. He was born with a cesarean section at term with a birth weight of 2.2 kg but without been told had intrauterine growth retardation during pregnancy period. The boy was purple after birth and improved after stimulation, but had feeding difficulties, so the boy was subsequent admission to the neonatal intensive care unit for weight management. Feeding difficulties and developmental delay were observed in infancy. He only sat at 10 months, walked at 18 months and spoke his first sentences at 2 years old. Growth delay persisted (current weight is 31.5 kg [P3–10], height 146.8 cm [P10–25], body mass index 14.62 kg/m2 [P3–5], and occipitofrontal circumference 52 cm [− 0.14SD] according to Chinese domestic data. Mild clinodactyly of both hands and abnormal facial features including wide-spaced eyes, prominent nasal bridge continuing to forehead, abnormal teething and micrognathia were noticed (Fig. 1 & Table 1). There was no history of seizures. Brain magnetic resonance image (MRI), EEG, X-ray of chest and spine, echocardiography and abdominal ultrasound were normal. Molecular karyotyping was normal.

a & b Facial features of the proband including wide-spaced eyes, prominent nasal bridge continuing to forehead, abnormal teething and micrognathia. c Mild clinodactyly was noticed on both hands. d Sanger confirmation of the truncating variants detected in the pedigree. e Sketch map of NSD2 gene domains and variants location of previously reported cases. (PWWP: Pro-Trp-Trp-Pro conserved motif; HMG: High mobility group box; PHD: Plant Homeodomain finger; AWS: associated with SET domain; SET: suppressor of variegation, enhancer of zeste, and Trithorax domain)
Ethical approval for this study was obtained from the ethics committee of Shanghai Children’s Medical Center, Shanghai Jiaotong University School of Medicine. Written informed consent permit to pusblish was obtained from the proband’s parents.
Whole exome capture was performed with Agilent SureSelect V6 enrichment capture kit (Agilent Technologies, Inc., Woburn, MA, U.S.) according to the manufacturer’s instructions. The captured library was then sequenced on an Illumina HiSeq 2500 System (Illumina, Inc., San Diego, CA, U.S.). Raw sequencing data was process as previously described [10]. All detected variants were analyzed on the TGex (Translational Genomics Expert) platform featuring with the VarElect scoring system [11]. An insertion variant c.4029_4030insAA leading to frameshift mutation (p.Glu1344Lysfs*49) on NSD2 was detected and thought to have high probability as a candidate mutation.
The primers for amplification of the NSD2 gene (NM_ 133,330.2) were designed using UCSC Exon Primer online software (http://genome.ucsc.edu/index.html) and synthesized. The primer sequences for the truncating variant to be confirmed were forward5’-agtttgtctgcccgtcctgt-3′ and reverse 5′-TGAGGATGGCTCAGTGGTG-3′.
The target fragments were amplified from the patients as well as his parents using polymerase chain reaction (Takara Biotechnology, Co., Ltd., Dalian, China). The PCR products were sequenced using an ABI3730XL sequencer (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, U.S.) with the forward and reverse primers. The sequence data were analyzed using Mutation Surveyor® software Version 4.0.4 (SoftGenetics, LLC). The truncating variant has been confirmed as de novo variants in the proband.
Discussion and conclusion
Critical pathogenic regions responsible for microdeletion syndromes are mapped by overlapping deletions and could in some cases help to reveal a single causative gene for the syndrome. For patients with WHS, NSD2 is the possible pathogenic gene located in the overlapping critical region previously defined by clinical and genomic information [12]. NSD2, nuclear receptor-binding SET domain-protein 2 (also known as WHSC1, Wolf-Hirschhorn syndrome candidate 1), is a SET domain histone methyltransferase, responsible for the methylation of HEK36. The importance of histone modification in brain development was gradually recognized as many pathogenic allele in genes encoding histone modification components was identified [13]. The histone substrate specificity of NSD2 encoded protein may explain differences in clinical phenotypes in related patients [14]. NSD2 gene is thought to be intolerant of loss of function variants (pLi = 1.00). Sequencing data from large cohorts of developmental delay, autism, and congenial cardiac problems also revealed several de novo variants in NSD2 that may be related to disease [15]. Case report with de novo truncating mutations on NSD2 with very detailed clinical information further supported that loss-of-function variants in NSD2 likely contributes to atypical or part of the clinical spectrum of WHS [7,8,9]. Herein, we reported another patient with disease-related frameshift mutation on NSD2 and reviewed clinical manifestation of previously reported patients.
Minimal diagnostic criteria for WHS have been proposed by including typical facial appearance, mental retardation, congenital hypotonia, growth delay and seizures [6]. The “Greek warrior helmet” appearance of the nose (wide bridge of the nose continuing to the forehead) is the most recognizable facial features for patients with WHS, but might become less evident after puberty. Our patient was considered for genetic testing initially due to observation of obvious abnormal appearance of the nose. Reviewing four reported cases and our case, their facial features are less recognizable comparing to typical 4p16.3 deletion WHS patients. Developmental delay or intellectual disability was observed in all four detailed reported cases. Battaglia et al. found that the degree of intellectual disability varies in patients with typical 4p deletion caused WHS [2]. The severity of intellectual disability seems to be much milder in reviewed and our patient. Seizures occur in 90–100% of children with WHS with a peak onset incidence around six to 12 months and represent one of the greatest problems in the clinical management of WHS and they act as an independent prognostic factor for the final degree of intellectual disability. Interestingly, none of the five patients reviewed with de novo truncating NSD2 variants had epileptic symptoms, indicating that other genes located within 4p16.3 region might be responsible for seizure pathogenesis. LETM1 and additional genes residing on 4pter distally to it have been suggested as candidate genes for seizure disorders in WHS patients in a comorbidity model of pathogenesis [16]. Thus, patients with truncating variants in NSD2 and without seizure attack before the age of one might have better intellectual performance. Skeletal anomalies were observed in our patients. He has mild clinodactyly on both hands. The 16 month-old boy reported by Lozier et al. also manifest clinodactyly of fifth fingers of the left hand [9]. Hand malformation is a much milder symptom compared to other skeletal anomalies like kyphosis/scoliosis with malformed vertebral bodies, accessory or fused ribs, clubfeet, and split hand found in 60–70% of WHS patients. Thus, we believe that truncating variants on NSD2 gene will lead to atypical clinical manifestations of WHS. Those patients may not have typical facial features and epileptic symptoms. The proposed minimal diagnostic criteria may not be suitable for these patients, and the optimal diagnosis criteria for them should including identification of null variants on NSD2 gene by genetic testing.
In conclusion, we identified a de novo truncating variant on NSD2 gene in a patient with mild phenotypic spectrum of WHS, further supported the pathogenesis of null variants in NSD2 in syndromic intellectual disability and developmental delay and these variants lead to a mild form of WHS.
Availability of data and materials
The datasets (whole-exome sequencing and Sanger sequencing files) used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- NSD2:
-
Nuclear receptor-binding SET domain-protein 2
- pLi:
-
Probability of loss-of function variants intolerance
- WHS:
-
Wolf-Hirschhorn syndrome
- WHSCR:
-
WHS critical region
References
Battaglia A, Carey JC, South ST. Wolf-Hirschhorn Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews. Seattle: University of Washington; 2015.
Battaglia A, Carey JC, South ST. Wolf-Hirschhorn syndrome: a review and update. Am J Med Genet C Semin Med Genet. 2015;169(3):216–23.
Battaglia A, Filippi T, Carey JC. Update on the clinical features and natural history of wolf-Hirschhorn (4p-) syndrome: experience with 87 patients and recommendations for routine health supervision. Am J Med Genet C Semin Med Genet. 2008;148C(4):246–51.
Zollino M, Murdolo M, Marangi G, Pecile V, Galasso C, Mazzanti L, Neri G. On the nosology and pathogenesis of wolf-Hirschhorn syndrome: genotype-phenotype correlation analysis of 80 patients and literature review. Am J Med Genet C Semin Med Genet. 2008;148C(4):257–69.
Wright TJ, Ricke DO, Denison K, Abmayr S, Cotter PD, Hirschhorn K, Keinänen M, McDonald-McGinn D, Somer M, Spinner N, Yang-Feng T, Zackai E, Altherr MR. A transcript map of the newly defined 165 kb wolf-Hirschhorn syndrome critical region. Hum Mol Genet. 1997;6(2):317–24.
Zollino M, Lecce R, Fischetto R, Murdolo M, Faravelli F, Selicorni A, Buttè C, Memo L, Capovilla G, Neri G. Mapping the wolf-Hirschhorn syndrome phenotype outside the currently accepted WHS critical region and defining a new critical region, WHSCR-2. Am J Hum Genet. 2003;72(3):590–7.
Derar N, Al-Hassnan ZN, Al-Owain M, Monies D, Abouelhoda M, Meyer BF, Moghrabi N, Alkuraya FS. De novo truncating variants in WHSC1 recapitulate the wolf-Hirschhorn (4p16.3 microdeletion) syndrome phenotype. Genet Med. 2019;21(1):185–8.
Boczek NJ, Lahner CA, Nguyen TM, Ferber MJ, Hasadsri L, Thorland EC, Niu Z, Gavrilova RH. Developmental delay and failure to thrive associated with a loss-of-function variant in WHSC1 (NSD2). Am J Med Genet A. 2018;176(12):2798–802.
Lozier ER, Konovalov FA, Kanivets IV, Pyankov DV, Koshkin PA, Baleva LS, Sipyagina AE, Yakusheva EN, Kuchina AE, Korostelev SA. De novo nonsense mutation in WHSC1 (NSD2) in patient with intellectual disability and dysmorphic features. J Hum Genet. 2018;63(8):919–22.
Yao R, Yu T, Xu Y, Li G, Yin L, Zhou Y, Wang J, Yan Z. Concurrent somatic KRAS mutation and germline 10q22.3-q23.2 deletion in a patient with juvenile myelomonocytic leukemia, developmental delay, and multiple malformations: a case report. BMC Med Genet. 2018;11(1):60.
Stelzer G, Plaschkes I, Oz-Levi D, Alkelai A, Olender T, Zimmerman S, Twik M, Belinky F, Fishilevich S, Nudel R, Guan-Golan Y, Warshawsky D, Dahary D, Kohn A, Mazor Y, Kaplan S, Iny Stein T, Baris HN, Rappaport N, Safran M, Lancet D. VarElect: the phenotype-based variation prioritizer of the GeneCards suite. BMC Genomics. 2016;17(Suppl 2):444.
Hannes F, Hammond P, Quarrell O, Fryns JP, Devriendt K, Vermeesch JR. A microdeletion proximal of the critical deletion region is associated with mild wolf-Hirschhorn syndrome. Am J Med Genet A. 2012;158A(5):996–1004.
Faundes V, Newman WG, Bernardini L, Canham N, Clayton-Smith J, Dallapiccola B, Davies SJ, Demos MK, Goldman A, Gill H, Horton R, et al. Histone lysine Methylases and demethylases in the landscape of human developmental disorders. Am J Hum Genet. 2018;102(1):175–87.
Douglas J, Coleman K, Tatton-Brown K, Hughes HE, Temple IK, Cole TR, Rahman N, Childhood Overgrowth Collaboration. Evaluation of NSD2 and NSD3 in overgrowth syndromes. Eur J Hum Genet. 2005;13(2):150–3.
Zollino M, Orteschi D, Ruiter M, Pfundt R, Steindl K, Cafiero C, Ricciardi S, Contaldo I, Chieffo D, Ranalli D, Acquafondata C, Murdolo M, Marangi G, Asaro A, Battaglia D. Unusual 4p16.3 deletions suggest an additional chromosome region for the wolf-Hirschhorn syndrome-associated seizures disorder. Epilepsia. 2014;55(6):849–57.
Monies D, Abouelhoda M, AlSayed M, Alhassnan Z, Alotaibi M, Kayyali H, Al-Owain M, Shah A, Rahbeeni Z, Al-Muhaizea MA, et al. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Hum Genet. 2017;136(8):921–39.
Acknowledgments
We would like to express our sincere gratitude to our patient and his family for their cooperation.
Funding
The study was supported by the Chinese National Natural Science Foundation (81602868); Shanghai Science and Technology Commission of Shanghai Municipality (17411965300, 17XD1402800); Shanghai Municipal Commission of Health and Family Planning (20164Y0095). These funding bodies contributed to design of the study and collection, analysis and interpretation of data.
Author information
Authors and Affiliations
Contributions
RY and FJ designed and organized the study. YJ, QL, and HS sampled the family members and acquired the clinical data. RY and JW carried out the molecular genetic testing. RY and WJ analyzed and interpreted the genetic testing and clinical data. RY wrote the manuscript, which was then edited by FJ and YJ. All authors have read and approved the final version of the manuscript submitted by RY.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Committee on Ethics of the Shanghai Children’s Medical Center (SCMCIRB-K2016013) and was performed in accordance with the Declaration of Helsinki.
Consent for publication
Written informed consent was obtained from the patient’s father for publication of this research.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jiang, Y., Sun, H., Lin, Q. et al. De novo truncating variant in NSD2gene leading to atypical Wolf-Hirschhorn syndrome phenotype. BMC Med Genet 20, 134 (2019). https://doi.org/10.1186/s12881-019-0863-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-019-0863-2