
Abstract
Background
Osteodysplasia of the oral and maxillofacial bone is generally accompanied by systemic bone abnormalities (such as short stature, joint contracture) or other systemic abnormalities (such as renal, dermatological, cardiovascular, optic, or hearing disorders). However, it does not always present this way. Recent reports have suggested that genome-wide sequencing is an effective method for identifying rare or new disorders. Here, we performed whole-exome sequencing (WES) in a patient with a unique form of acquired, local osteodysplasia of the oral and maxillofacial region.
Case presentation
A 46-year-old woman presented to our hospital with the complaint of gradually moving mandibular teeth (for 6 months), changing facial appearance, and acquired osteolysis of the oral and maxillofacial bones, showing mandibular hypoplasia without family history. Upon skeletal examination, there were no abnormal findings outside of the oral and maxillofacial area; the patient had a height of 157 cm and bone mineral density (according to dual energy x-ray absorptiometry) of 90%. Results of blood and urine tests, including evaluation of bone metabolism markers and neurological and cardiovascular examinations, were normal. We performed WES of genomic DNA extracted from the blood of this patient and her mother, who did not have the disease, as a negative control. We identified 83 new missense variants in the patient, not detected in her mother, including a candidate single nucleotide variant in exon 14 of PCNT (pericentrin). Critical homozygous or compound heterozygous variants in PCNT are a known cause of microcephalic osteodysplastic primordial dwarfism type II accompanied by mandibular hypoplasia, which is similar to the maxillofacial phenotype in this patient.
Conclusions
Protein simulations performed using Polymorphism Phenotyping v2 and Combined Annotation Dependent Depletion software indicated that this missense variant is likely to disrupt the PCNT protein structure. These results suggest that this is a new form of osteolysis related to this PCNT variant.
Similar content being viewed by others


Background
Osteodysplasia of the oral and maxillofacial bone is generally accompanied by systemic metabolic bone disease or other systemic abnormalities, such as cardiac malformations or neurological disorders [1,2,3]. Acquired local osteodysplasia without metabolic disease is usually accompanied by the presence of abnormal soft tissues, including malignant tumors [3]. However, acquired local osteodysplasia is rarely reported.
The relative affordability and accessibility of genome-wide sequencing have facilitated the development of family-based genomic analysis, resulting in an explosion of gene discovery and diagnosis of rare diseases [4]. However, for many congenital malformations, identification of the causative mutation by whole-genome sequencing or whole-exome sequencing (WES) has been challenging [5]. Nonetheless, recent studies have identified specific gene variants in patients with congenitally acquired skeletal disorders, and genome-wide sequencing is a potent technique for the identification of variants implicated in unknown disorders [6].
Here, we performed WES in a patient with a potentially unknown skeletal disorder involving oral and maxillofacial acquired local osteodysplasia without metabolic disease or soft tissue around the bone lesions.
Case presentation
Case history
In 2015, a 46-year-old woman presented to the Department of Oral and Maxillofacial Surgery of Tokai University Oiso Hospital, Kanagawa, Japan, with the complaint of gradually moving mandibular teeth (for 6 months) and changing facial appearance. She had been referred by her family dentist, who had noticed the early stages of mandibular bone loss 9 years prior. She had received no treatment other than periodontal therapy performed by her dentist.
Family history
The patient was the youngest of two children. Her mother and father were 32 and 36 years old, respectively, at the time of her birth. Her father died in an accident at the age of 40. Her sister did not exhibit similar symptoms or present with facial malformation.
Medical history
Birth weight and intelligence level were normal. The patient’s history was remarkable for 10 episodes of bacterial meningitis, which occurred between 31 and 39 years of age. She was premenopausal at presentation.
Physical findings
We consulted an orthopedic surgeon and genetic physician with the intent of performing a comprehensive screening for systemic disorders. Upon skeletal examination, there were no abnormal findings outside of the oral and maxillofacial area; the patient had a height of 157 cm and bone mineral density (according to dual energy x-ray absorptiometry) of 90%. Results of blood and urine tests for bone metabolism markers, including calcium (9.8 mg/dL), phosphorous (4.4 mg/dL), alkaline phosphatase (284 IU), 25-OH-Vitamin D (55 pg/mL), and collagen cross-linked N-telopeptide (18.4 nmol), as well as neurological and cardiovascular examinations, were normal.
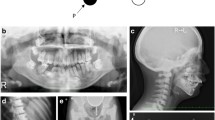
Extraoral examination revealed mandibular hypoplasia but no asymmetry (Fig. 1a). Intraoral examination revealed an anterior open bite (inter-incisor distance: 23 mm) and no crowding of the mandibular teeth. There were no abnormal findings pertaining to the oral mucosa (Fig. 1b).

Patient imaging data. a Photographs from extraoral examination reveal mandibular hypoplasia but no asymmetry. b Photographs from intraoral examination reveal an anterior open bite (inter-incisor distance: 23 mm) and early contact of the second molars. The upper teeth are crowded without a cleft palate; the lower teeth are not crowded
Radiographic findings
Posteroanterior and lateral radiographs of the skull did not reveal any abnormal morphology of the head or facial height and width (Fig. 2a). Panoramic radiographic images and computed tomography scans showed axial and coronal thinning of the alveolar bone, anterior wall of the maxillary sinus in the maxilla, and the entire mandibular (condyle, angle, body, and alveolar) bone (Figs. 2b and 3). Magnetic resonance imaging did not show any soft tissue masses in the maxillofacial area (data not shown). Technetium (99mTc) bone scintigraphy showed tracer uptake in the maxillary and mandibular bones (Fig. 4).

X-ray images. a Posteroanterior and lateral skull views do not reveal morphological abnormalities of the head or face height and width. b A panoramic radiographic image shows axial and coronal thinning of the alveolar bone in the maxilla and the entire mandibular (condyle, angle, body, and alveolar) bone

Computed tomography imaging. Imaging shows axial and coronal thinness of the alveolar bone, anterior wall of the maxillary sinus in the maxilla, and the entire mandibular (condyle, angle, body, and alveolar) bone

99mTc bone scintigraphy shows tracer uptake in the maxillary and mandibular bones
DNA extraction, library preparation, and sequencing
Genomic DNA was extracted from 100 μL of whole blood from the patient and her mother (as a negative control) using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) following the manufacturer’s recommendations. Initial DNA sample quality assessment, library preparation, and sequencing were conducted by GENEWIZ, Inc. (South Plainfield, NJ, USA). The SureSelectXT Target Enrichment System for Illumina Paired-End Multiplexed Sequencing Library and SureSelect Human All Exon V6 bait library (Agilent, Santa Clara, CA, USA) were used for target enrichment and DNA library preparation following the manufacturer’s recommendations.
The sequencing library was loaded onto an Illumina HiSeq instrument (San Diego, CA, USA) according to the manufacturer’s instructions. Raw sequence data (.bcl files) generated from the Illumina HiSeq instrument were converted into fastq files and de-multiplexed using bcl2fastq software 2.17 (Illumina). One mismatch was allowed for index sequence identification. The data of whole exome sequence was assigned on The DNA Data Bank of Japan Center (NBDC number: hum0190) [7].
The average coverage depth of the entire panel was 137×, and 99.9% of targeted bases were covered by sequence reads at a depth of at least 20 × .
Candidate gene approach and gene annotation
Sequence reads were aligned using BWA (version 0.7.12) [doi:https://doi.org/10.1093/bioinformatics/btp324]. Local realignment and base quality recalibration was performed using GATK (version 3.5) [doi: https://doi.org/10.1101/g.107524.110]. Variant calling was performed with SAMtools (version 1.3.1) [doi.org/10.1093/bioinformatics/btp352] [doi.org/10.1093/bioinformatics/btr509]. Variants were annotated using the ANNOVAR tool [doi: https://doi.org/10.1093/nar/gkq603].
Only non-synonymous sequence differences between the mother and patient in the sequenced genes were analyzed. Candidate gene sequencing revealed 83 heterozygous variants in 79 genes, which were confirmed by Sanger sequencing (Table 1). These single-nucleotide variants (SNVs) were not present in any of the queried population databases [(Integrative Japanese Genome Variation Databas (iJGVD), The International Genome Sample Resource and Providing ongoing support for the 1000 Genomes Project data (1000 Genomes), The Exome Aggregation Consortium (ExAC), snp138, Human Genetic Variation Database (HGVD), and The Genome Aggregation Database (gnomAD)]. Among the 83 heterozygous variants, 12 SNVs in 11 genes (AHNAK, CCDC15, COBL, DCAF5, MCF2L, NSRP1, NSRP1, PCNT, RMDN3, and TTYH2) were considered to be “probably damaging” according to Polymorphism Phenotyping v2 (PolyPhen-2) (http://genetics.bwh.harvard.edu/pph2/) and Combined Annotation Dependent Depletion (CADD) [https://cadd.gs.washington.edu/]. These databases predict the potential effect of an amino acid substitution on the structure and function of a human protein using straightforward physical and comparative considerations [8].
The 11 genes with probably damaging SNVs were analyzed using the Genecards® human gene database version 4.5 [https://www.genecards.org/]. AHNAK may be involved in diverse processes such as blood-brain barrier formation, cell structure and migration, cardiac calcium channel regulation, and tumor metastasis [9]. COBL may play a role in maintaining intestinal homeostasis [10]. NSRP1 is an mRNA binding protein that has not been associated with any clinical features [11]. The molecular and clinical functions of CCDC15, DCAF5, and RMDN3 have not been clarified according to Genecards®. MCF2L is related to the Rho/Rac signaling pathways, which play key roles in cell proliferation, migration, and motility, including in cancer metastasis [12]. Among the 11 genes, only PCNT is expressed in the cortex and skeletal muscle. We speculate that the PCNT variant (chr21 47783755: NM_006031, exon14, c.2515 T > C, p.839C > R; NM_001315529, exon 14, c.2161 T > C, p.721C > R) in the patient may affect the cell division of osteoblasts or osteoclasts and bone homeostasis in the oral and maxillofacial area. This differs from the result of PCNT-null disorders (Fig. 5a). Thus, we considered PCNT a candidate gene for this disorder because of its high mRNA expression in the cortex and skeletal muscle and the lack of clinical reports for AHNAK, CCDC15, COBL, DCAF5, MCF2L, NSRP1, NSRP1, RMDN3, and TTYH2. Notably, p.C721R/p.C839R of PCNT has not yet been reported as a candidate variant for skeletal disorders in the Nosology and Classification of Genetic Skeletal Disorders, which serves as a “master” list of genetic disorders of the skeleton to facilitate diagnosis and to help delineate variants or newly recognized conditions [13].

Missense variant present in exon 14. The variant was determined to be PCNT: NM_006031, exon 14, c.T2515C, p.C839R; NM_001315529, exon 14, c.T2161C, p.C721R. a Sanger sequencing results for the patient and her mother. b Mutation map of two isoforms (transcript variants)
Treatment and follow-up
Because this osteolytic disorder had not been previously reported and no specific treatment was indicated, the patient was placed under observation with periodontal maintenance. No progression has been noted in the 2.5 years since diagnosis.
Discussion and conclusions
Osteolysis in the oral and maxillofacial bones can be a phenotype of various systemic disorders, calcium and phosphorus disorders, hyperparathyroidism, hypoparathyroidism, osteomalacia, osteopenia, osteoporosis, Paget’s disease, and vitamin D deficiency [1, 3, 6, 13, 14]. Congenital osteolysis is often hereditary and accompanied by disturbances in bone metabolism [14]. Major osteolysis can lead to skeletal malformations, such as short stature. Although familial osteodysplasia localized in the mandible was reported in four of six siblings (13 to 23 years old) in 1972, none of the patients had any offspring, and the genetic characteristics of this disease were not investigated [15].
Potential differential diagnoses of osteodysplasia syndromes, which involve bone anomalies in the oral and maxillofacial region, are shown in Table 2 [6, 16]. Gorham’s disease is an acquired condition characterized by local or massive osteolysis that can involve the oral and maxillofacial bones. In this disease, the osteolytic region is often accompanied by soft hemangiomatous or lymphangiomatous tissue [6]. In our patient, although osteolysis was shown to be localized in the oral and maxillofacial region, soft tissue did not appear around the bone lesions. Hence, we were not able to make a diagnosis based on clinical features. Therefore, we performed WES in the patient and her mother, who did not have a bone disorder, and sought to identify gene variants in known skeletal disorder genes and to clarify the genetic basis of this maxillofacial osteolysis.
PCNT is one of the calmodulin-binding proteins expressed in the centrosome. PCNT (< 370 kDa) contains a series of coiled-coil domains and localizes specifically to the centrosomes throughout the cell cycle [17]. The protein interacts with the microtubule nucleation component gamma-tubulin and is likely important for normal functioning of the centrosomes, cytoskeleton, and cell-cycle progression. Two transcript variants encoding different isoforms of this gene are annotated in the RefSeq database: NM_001315529.1 and NM_006031.5 [https://www.ncbi.nlm.nih.gov/nuccore/NM_001315529.1 and https://www.ncbi.nlm.nih.gov/nuccore/NM_006031.5]. The domain structure and function around p.C721R/p.C839R have not been determined.
Critical mutations such as homozygous or compound heterozygous mutations in PCNT are a known cause of microcephalic osteodysplastic primordial dwarfism type II (MOPD II; Mendelian Inheritance in Man: 210720). MOPD II is characterized by birth weight (14.3 ± 7.7 SD below the population mean and head circumference 8.5 ± 2.1 SD below the population mean, as well as a variety of associated systemic bone and dental anomalies, but there are not any actual cut-off values for diagnosis [18,19,20]. Our patient’s features differ from those of individuals with MOPD II. However, as our patient has mandibular hypoplasia (Fig. 1a), we speculate that this phenotype was secondarily caused by jaw osteolysis, similar to the phenotypes of MOPD II patients with micrognathia and retrognathia [18,19,20]. Genetic data from 25 German MOPD II patients, including three families of Turkish origin, were used in the first mutational analysis of PCNT, which revealed homozygous and compound heterozygous PCNT-null mutations (four splice-site mutations, two small insertions, 10 small deletions, and one exon deletion) [20]. In addition, one Colombian MOPD II patient (nonsense mutation, c.C1468T, in exon 10) and members of one Chinese MOPD II family (small deletions in exons 30 and 41) had apparently homozygous null mutations [21, 22]. PCNT-null fibroblast cells derived from MOPD II patients show that a loss of PCNT function induces abnormal mitotic morphology; however, the pathogenic effect of PCNT variants in skeletal disorders remains unclear [20].
The PCNT protein has two transcriptional isoforms in humans and mice: the full-length pericentrin protein (isoform 1) and an alternatively spliced form that lacks the N-terminal amino acids (isoform 2), the structure of which has not been determined (Fig. 5b) [23]. However, the molecular mobility of each isoform has not been determined in humans. In vitro and in vivo assays are necessary to identify the functional effect of p.C721R/p.C839R variant.
Bone biopsy is a useful tool for identifying the mechanism of skeletal disorders and determining treatment. Although bisphosphonates and drugs for primary diseases are used in the treatment of systemic bone disease [14, 24, 25], local osteolysis does not require particular therapy; however, patients often need reconstructive surgery [1, 3, 13, 26]. In the patient described herein, we did not perform a biopsy due to the risk of fracture. Thus, this patient was not diagnosed with a novel disorder, and we were unable to perform curative treatment. We consider that dental infection is a serious risk factor for the promotion of osteolysis in this patient and ultimately opted for conservative treatment, with the patient undergoing dental maintenance. Intensive follow up including X ray images if necessary and panoramic X ray is performed once every 3 months. If a mandibular fracture were to appear, reconstructive surgery would be necessary.
The WES results presented in this study suggest that the osteolysis in this patient represents a new disease related to the presence of a variant in PCNT. Further investigations are required to determine the function of PCNT and identify the mechanism behind acquired local bone osteolysis.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- MOPD II:
-
Microcephalic osteodysplastic primordial dwarfism type II
- WES:
-
Whole exome sequencing
References
Canalis E, Zanotti S. Hajdu-Cheney syndrome: a review. Orphanet J Rare Dis. 2014;9:200.
Luisa B, Valerie CD, Christine H, Ralph L, Geert M, Stefan M, Gen N, Luca S, Ravi S, David S, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A. 2015;167(12):2869–92.
Renacci RM, Bartolotta RJ. Gorham disease: lymphangiomatosis with massive osteolysis. Clin Imaging. 2017;41:83–5.
Wright CF, McRae JF, Clayton S, Gallone G, Aitken S, FitzGerald TW, Jones P, Prigmore E, Rajan D, Lord J, et al. Making new genetic diagnoses with old data: iterative reanalysis and reporting from genome-wide data in 1,133 families with developmental disorders. Genet Med. 2018;20:1216–23.
Hassan TM, Marie-Noel W, LA E, HL B. Causes of congenital malformations. Birth Defects Res. 2018;110(2):87–91.
Bonafe L, Cormier-Daire V, Hall C, Lachman R, Mortier G, Mundlos S, Nishimura G, Sangiorgi L, Savarirayan R, Sillence D, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A. 2015;167a(12):2869–92.
Kodama Y, Mashima J, Kosuge T, Katayama T, Fujisawa T, Kaminuma E, Ogasawara O, Okubo K, Takagi T, Nakamura Y. The DDBJ Japanese genotype-phenotype archive for genetic and phenotypic human data. Nucleic Acids Res. 2015;43(Database issue):D18–22.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Sohn M, Shin S, Yoo JY, Goh Y, Lee IH, Bae YS. Ahnak promotes tumor metastasis through transforming growth factor-beta-mediated epithelial-mesenchymal transition. Sci Rep. 2018;8(1):14379.
Grega-Larson NE, Crawley SW, Erwin AL, Tyska MJ. Cordon bleu promotes the assembly of brush border microvilli. Mol Biol Cell. 2015;26(21):3803–15.
Kim YD, Lee JY, Oh KM, Araki M, Araki K, Yamamura K, Jun CD. NSrp70 is a novel nuclear speckle-related protein that modulates alternative pre-mRNA splicing in vivo. Nucleic Acids Res. 2011;39(10):4300–14.
Ungefroren H, Witte D, Lehnert H. The role of small GTPases of the rho/Rac family in TGF-beta-induced EMT and cell motility in cancer. Dev Dyn. 2018;247(3):451–61.
Flannery O, Smith K. Osteolysis with secondary arthritis of the scaphotrapeziotrapezoid joint in Hajdu-Cheney syndrome: a case report. Hand Surg. 2014;19(1):117–8.
Glendenning P, Chubb SAP, Vasikaran S. Clinical utility of bone turnover markers in the management of common metabolic bone diseases in adults. Clin Chim Acta. 2018;481:161–70.
Anderson LG, Cook AJ, Coccaro PJ, Coro CJ, Bosma JF. Familial osteodysplasia. JAMA. 1972;220(13):1687–93.
Asha ML, Jose AA, Babshet M, Bajoria AA, Naveen S. Vanishing bone disease of the mandible - a case report. Gerodontology. 2013;30(4):296–302.
Flory MR, Davis TN. The centrosomal proteins pericentrin and kendrin are encoded by alternatively spliced products of one gene. Genomics. 2003;82(3):401–5.
BM G, HB D, MR N, LC B, OJ M, RJ F. Do some patients with seckel syndrome have hematological problems and/or chromosome breakage? Am J Med Genet. 1987;27(3):645–9.
HJ G, Christina F, SC I, PR M, TK I. Majewski osteodysplastic primordial dwarfism type II (MOPD II): natural history and clinical findings. Am J Med Genet A. 2004;130A(1):55–72.
Rauch A, Thiel CT, Schindler D, Wick U, Crow YJ, Ekici AB, van Essen AJ, Goecke TO, Al-Gazali L, Chrzanowska KH, et al. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science. 2008;319(5864):816–9.
Li F-F, Wang X-D, Zhu M-W, Lou Z-H, Zhang Q, Zhu C-Y, Feng H-L, Lin Z-G, Liu S-L. Identification of two novel critical mutations in PCNT gene resulting in microcephalic osteodysplastic primordial dwarfism type II associated with multiple intracranial aneurysms. Metab Brain Dis. 2015;30(6):1387–94.
Pachajoa H, Ruiz-Botero F, Isaza C. A new mutation of the PCNT gene in a Colombian patient with microcephalic osteodysplastic primordial dwarfism type II: a case report. J Med Case Rep. 2014;8(1):191.
Miyoshi K, Asanuma M, Miyazaki I, Matsuzaki S, Tohyama M, Ogawa N. Characterization of pericentrin isoforms in vivo. Biochem Biophys Res Commun. 2006;351(3):745–9.
Patel S, Lyons AR, Hosking DJ. Drugs used in the treatment of metabolic bone disease. Clinical pharmacology and therapeutic use. Drugs. 1993;46(4):594–617.
Peters J, Robertson A, Godavitarne C, Rogers B. Metabolic bone disease. Orthop Trauma. 2017;31(5):306–11.
Song HK, Sohn YB, Choi YJ, Chung YS, Jang JH. A case report of pycnodysostosis with atypical femur fracture diagnosed by next-generation sequencing of candidate genes. Medicine (Baltimore). 2017;96(12):e6367.
Acknowledgements
The authors thank Genewiz Japan for next-generation sequencing and Hideki Hayashi (Department of Bioinformatics, Support Center for Medical Research and Education, Tokai University) for providing bioinformatics support. We also would like to thank Editage (www.editage.jp) for English language editing.
Funding
This work was supported by Grants-in-Aid for Young Scientists (B) (KAKENHI Grant Number 16 K20614) from the Japan Society of the Promotion of Science (JSPS) for the design of the study and collection, analysis, and interpretation of data.
Author information
Authors and Affiliations
Contributions
KA, RK, YO, and MK wrote the manuscript, participated in the analysis of data, and edited images. KH, MT, SY, and GN performed the physical examination. HY, MU, and YO provided guidance in patient management and participated in manuscript preparation. All authors participated in manuscript revision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. The ethics committee of Tokai University Hospital approved this study (16I-31).
Consent for publication
We confirm that the patient and patients’ parent have given their written consents for the case report to be published, including their medical data and images.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Aoyama, Ki., Kimura, M., Yamazaki, H. et al. New PCNT candidate missense variant in a patient with oral and maxillofacial osteodysplasia: a case report. BMC Med Genet 20, 126 (2019). https://doi.org/10.1186/s12881-019-0858-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-019-0858-z