
Abstract
Background
Population structure is an important factor in the genetic association studies but often remains underexplored for many human populations. We identified exome variants in 39 Siberian Caucasian individuals from Novosibirsk, Russia and compared their genetic allele frequencies with European populations from 1000 Genomes Project.
Methods
The study participants were from Novosibirsk and represented people with monogenic diabetes, healthy individuals and a cohort from the tick-borne encephalitis study. Isolated DNA was enriched using Agilent SureSelect V5 kit and sequenced on Illumina HiSeq 4000 and genetic variants were identified using GATK pipeline. To estimate the patterns of the population structure we used PCA and ADMIXTURE analysis. Pharmocogenetically and medically important variants were annotated based on PharmGKB and ClinVar databases.
Results
The analysis identified low, but highly significant population differentiation attributed to numerous loci between the Siberian Caucasian population and other European population samples as well as a higher proportion of the Finnish genetic component in the studied sample. The medical and pharmacogenetic annotation of highly significantly differentiated variants between the Novosibirsk and the combined European populations revealed a number of important genetic polymorphisms located in such genes as FCGR3B, TYR, OCA2, FABP1, CHEK2 and SLC4A1.
Conclusions
The study reports for the first time an exome-wide comparison of a population from Russia with European samples and emphasizes the importance of population studies with medical annotation of variants.
Similar content being viewed by others


Background
Population structure is a very important factor in medical genetic association studies which can compromise modern genomic methods not being properly accounted for. In Russia, population studies were mainly conducted using Y-chromosome or mitochondrial markers with the recent application of microarray methods [1,2,3] and did not allow to estimate the functional role of variants. Some recent phylogeographic studies used whole-genome sequencing with samples from Russia to elucidate history of migrations in Eurasia, but used small samples from diverse populations [4, 5]. In this study, we identified exome genetic variants for 39 individuals from Novosibirsk, Russia and compared them with the previously published genome-wide data and exomes of European populations from the 1000 Genomes Project to understand the level of the exome-wide divergence and the extent of the population stratification. The Novosibirsk population (NVSB) is of particular interest because it exhibits an example of a modern big city population affected by political and economic events of the twentieth century which changed the historical landscape of ethnic diversity of the former USSR territory through increasing urbanization, mass migration across the country and rapid demographic growth. In this study, we identified exome genetic variants for 39 individuals from Novosibirsk, Russia and compared them with the previously published genome-wide data and exomes of European populations from the 1000 Genomes Project to understand the level of the exome-wide divergence and the extent of the population stratification. Additionally, we tested allele frequency differences between our sample and combined European dataset for medically and pharmacogenetically important variants to identify loci which can be important for national studies.
Methods
The study participants (n = 39) were from Novosibirsk and represented people with monogenic diabetes (n = 10), healthy individuals (n = 7) and a cohort from the tick-borne encephalitis study (n = 22). The participants signed an informed consent and defined themselves as ethnic Russians. The ethnicity of the participants was additionally checked prior the analysis with data from 1000 Genomes Project and two samples identified as clear outliers (close to the Asian populations) were excluded from the analysis. Isolated DNA was enriched using Agilent SureSelect V5 kit in and sequenced on Illumina HiSeq 4000 with 150PE reads. After the quality control with Trimmomatic [6] the reads were aligned with BWA mem [7] to Hg19 reference genome and processed with SAMtools [8]. Single nucleotide variants (SNVs) and indels were identified using GATK [9] according to the GATK Best Practices workflow for germline variation with the sensitivity filter equal to 99.9. The resulted VCF file was combined with 1000 Genomes Project genotypes [10] using bcftools [11] merge and filtered with VCFtools [11] at maximum 10 missed genotypes (−-max-missing-count) keeping only biallelic sites.
We performed the analysis on the two levels: with the Finnish (FIN) population for population genetic analysis (PCA, ADMIXTURE, Fst) and without FIN population to test the allele frequency differences for clinically and pharmacogenetically important variants. The FIN population was excluded from the second analysis as the most divergent European population with unique history [12]. To reduce the influence of the tightly linked loci on the patterns of population structure we applied the linkage-disequilibrium pruning using PLINK V1.93 software (Table 1). To estimate the patterns of the population structure we used the Principal Component Analysis (PCA) realised in SNPrelate [13] with European (1000 Genomes Project) and previously published Russian Siberian populations [2]. The proportions of genetic ancestry between populations were estimated using ADMIXTURE [14] for K = 2–8 (Table 1) and tested using Cross Validation Error estimation (CVE). To estimate and test statistically the level of pairwise population differentiation (Fst, [15]) we used smartpca software of the EIGENSTRAT package [16].
We annotated the variants using ANNOVAR [17] and PharmGKB [18] databases and then tested medically (ClinVar, [19]) and pharmacogenetically relevant variants for the differences in allele frequencies between the NVSB population and the combined non-Finnish European (NFE) dataset with PLINK v1.93 [20] using 1 M permutations.
The average coverage of the studied exomes varied from 47.7X to 71.3X. In total, we identified 136,276 SNVs and 14,464 indels in the studied dataset. Merging with data from 1000 Genomes Project produced a dataset of overlapped variants consisted of 117,010 SNVs and 5989 indels.
Results
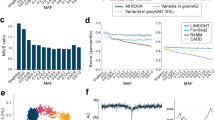
During the population genetic analysis, the first principal component accounted for 0.77% of the total variation and separated all the populations (Fig. 1a) except closely related American (CEU) and British (GBR). The second principal component accounted for 0.36% of the total variation and separated mostly Tuscan (TSI) and Spanish (IBS) samples. Novosibirsk population (NVSB) was placed between the Finnish (FIN) and CEU with GBR samples and was clearly distinguished from them. The Russian Siberian populations from a previous microarray-based study [2], represented by a similar Caucasian Siberian population (Russian_NSK) and partially isolated Siberian Starovers (Old Believers, Russian_STV) were not distinguished between each other and samples from our study (NVSB).

a Principal Component Analysis (Russian_NSK and Russian_STV are Russian from Novosibirsk and Siberian starovers respectively from [2]) b Observed and expected P-value distribution for allele frequency differences between NVSB and combined NFE sample c Results of the ADMIXTURE analysis for K = 2–5
In ADMIXTURE analysis, the lowest value of the Cross Validation Error was attributed to the K = 2, which captured the divergence of FIN from other European populations. NVSB demonstrated a higher proportion of the ancestral Finnish-related genetic component at K = 2 and at K = 3 relative to other populations. A new cluster (green) consisted of TSI and IBS appeared at K = 3 and then at an additional ancestral component emerged (K = 4) clearly separating NVSB (Fig. 1c, purple). Lastly, at K = 5, the IBS was separated from the rest of the samples.
The pairwise Fst values between all the populations except the CEU and GBR (P-value = 0.048) were highly significant (P-value < 1.1656e-11) albeit low (Fst = 0.002–0.013). The NVSB population demonstrated the highest level of differentiation with TSI (Fst = 0.009) and the lowest with GBR and CEU (Fst = 0.005). The results of the test for allele frequency differences between NVSB and NFE populations demonstrated pervasive inflation of the P-values attributed to numerous loci (Fig. 1b).
Among the 452 pharmocogenetically and 210 medically important variants we found 3 and 7 variants respectively (Table 2) which showed significant allele frequency differences between the NVSB and NFE population after the multiple testing correction (BH adjusted P-value < 0.05). The most significant differences in allele frequencies were attributed to such genes as FCGR3B, TYR, OCA2, FABP1 and SLC4A1 genes.
Discussion
In this study, we used an exome-wide dataset for the first time to study the population structure of the Caucasian Siberian population from a big Russian city Novosibirsk. The exome-wide survey of the Novosibirsk population demonstrated its genetic congruence with the previously published Russian dataset including the partially isolated Siberian Starovers regardless of the dramatic migration and demographic changes of the previous century. The Caucasian Novosibirsk population is quite homogeneous (Fig. 1a) and significantly differentiated from other European populations from 1000 Genomes Project demonstrating a relatively higher Finnish component which is presumably ancestral but not a result of recent migrations according to the ADMIXTURE results (Fig. 1c). This genetic differentiation although low in absolute Fst values should be taken into account during association studies. We identified 10 medically relevant SNVs with statistically significant allele differences between the NVSB and NFE populations including rs2241883 in FABP1 gene previously associated with polycystic syndrome [21] and toxicity of fenofibrate [22], rs1801274 variant in FCGR2A gene shown to be important for the efficiency of trastuzumab in breast neoplasms [23], the rare rs17879961 variant in CHEK2 gene reliably associated with predisposition to breast and colorectal cancer [24] and showed elevated frequency in NVSB. These variants should be studied in future on an expanded dataset with associated clinical data.
Conclusion
The study reports for the first time an exome-wide comparison of a population from Russia with European samples and emphasizes the importance of population studies with medical annotation of variants.
References
Kushniarevich A, Utevska O, Chuhryaeva M, Agdzhoyan A, Dibirova K, Uktveryte I, et al. Genetic Heritage of the Balto-Slavic Speaking Populations: A Synthesis of Autosomal, Mitochondrial and Y-Chromosomal Data. PLoS One. 2015;10(9):e0135820. Available from: https://doi.org/10.1371/journal.pone.0135820
Triska P, Chekanov N, Stepanov V, Khusnutdinova EK, Kumar GPA, Akhmetova V, et al. Between Lake Baikal and the Baltic Sea: genomic history of the gateway to Europe. BMC Genet. 2017;18(S1). Available from: https://doi.org/10.1186/s12863-017-0578-3
Khrunin AV, Khokhrin DV, Filippova IN, Esko T, Nelis M, Bebyakova NA, et al. A Genome-wide analysis of populations from European Russia reveals a new pole of genetic Diversity in northern Europe. PLoS One. 2013;8(3):1–9.
Wong EHM, Khrunin A, Nichols L, Pushkarev D, Khokhrin D, Verbenko D, et al. Reconstructing genetic history of Siberian and northeastern European populations. Genome Res. 2016;27(1):1–14. Available from:. https://doi.org/10.1101/gr.202945.115.
Mallick S, Li H, Lipson M, Mathieson I, Gymrek M, Racimo F, et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature . 2016 21;538:201. Available from: https://doi.org/10.1038/nature18964.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30(15):2114–2120. Available from: https://doi.org/10.1093/bioinformatics/btu170
Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics [Internet]. 2009;25(14):1754–1760. Available from: https://doi.org/10.1093/bioinformatics/btp324
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009;25(16):2078–2079. Available from: https://doi.org/10.1093/bioinformatics/btp352
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20(9):1297–1303. Available from: https://doi.org/10.1101/gr.107524.110
An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. Available from: https://doi.org/10.1038/nature11632
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics 2011;27(15):2156–2158. Available from: https://doi.org/10.1093/bioinformatics/btr330
Kääriäinen H, Muilu J, Perola M, Kristiansson K. Genetics in an isolated population like Finland: a different basis for genomic medicine? J Community Genet 2017;8(4):319–326. Available from: https://doi.org/10.1007/s12687-017-0318-4
Zheng X, Levine D, Shen J, Gogarten SM, Laurie C, Weir BS. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 2012;28(24):3326–3328. Available from: https://doi.org/10.1093/bioinformatics/bts606
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res 2009;19(9):1655–1664. Available from: https://doi.org/10.1101/gr.094052.109
Hudson RR, Slatkin M, Maddison WP. Estimation of levels of gene flow from DNA sequence data. Genetics. 1992;132(2):583 LP–589 Available from: http://www.genetics.org/content/132/2/583.abstract.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006;38(8):904–909. Available from: https://doi.org/10.1038/ng1847
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38(16):e164–e164. Available from: https://doi.org/10.1093/nar/gkq603
Hewett M. PharmGKB: the pharmacogenetics Knowledge Base. Nucleic Acids Res [Internet]. 2002;30(1):163–165. Available from: https://doi.org/10.1093/nar/30.1.163
Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2013;42(D1):D980–D985. Available from: https://doi.org/10.1093/nar/gkt1113
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-Genome association and population-based linkage analyses. Am J Hum Genet 2007;81(3):559–575. Available from: https://doi.org/10.1086/519795
Xue H, Zhao H, Liu X, Zhao Y, Chen Z-J, Ma J. Association of single-nucleotide polymorphisms rs2197076 and rs2241883 of FABP1 gene with polycystic ovary syndrome. J Assist Reprod Genet 2015;33(1):75–83. Available from: https://doi.org/10.1007/s10815-015-0626-8
Brouillette C, Bossé Y, Pérusse L, Gaudet D, Vohl M-C. Effect of liver fatty acid binding protein (FABP) T94A missense mutation on plasma lipoprotein responsiveness to treatment with fenofibrate. J Hum Genet. 2004;49(8). Available from: https://doi.org/10.1007/s10038-004-0171-2
Tamura K, Shimizu C, Hojo T, Akashi-Tanaka S, Kinoshita T, Yonemori K, et al. Fc R2A and 3A polymorphisms predict clinical outcome of trastuzumab in both neoadjuvant and metastatic settings in patients with HER2-positive breast cancer. Ann Oncol 2010;22(6):1302–1307. Available from: https://doi.org/10.1093/annonc/mdq585
Han F, Guo C, Liu L. The effect of CHEK2 variant I157T on Cancer susceptibility: evidence from a meta-analysis. DNA Cell Biol 2013;32(6):329–335. Available from: https://doi.org/10.1089/dna.2013.1970
Acknowledgments
We acknowledge A. Bielhin for English language editing of the manuscript.
Funding
This study and publication costs are funded by the grant from the Russian Science Foundation (project no. 16–15-00127).
Availability of data and materials
The dataset (individual genetic variants) analysed during the current study is available from the corresponding author on reasonable request following approval of the Institute of Cytology and Genetics (Siberian Branch, Russian Academy of Sciences) authorities and the Bioethics Committee.
About this supplement
This article has been published as part of BMC Medical Genetics Volume 20 Supplement 1, 2019: Selected articles from BGRS\SB-2018: medical genetics. The full contents of the supplement are available online at https://bmcmedgenet.biomedcentral.com/articles/supplements/volume-20-supplement-1.
Author information
Authors and Affiliations
Contributions
AY, NY and MV designed the study; AY performed data analysis; AY wrote the draft of the manuscript; all authors read, edited and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This research was approved by the Bioethics Committee of the Federal Research Centre Institute of Cytology and Genetics (Siberian Branch, Russian Academy of Sciences). All the participants signed an informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yurchenko, A.A., Yudin, N.S. & Voevoda, M.I. Exome-wide survey of the Siberian Caucasian population. BMC Med Genet 20 (Suppl 1), 51 (2019). https://doi.org/10.1186/s12881-019-0772-4
Published:
DOI: https://doi.org/10.1186/s12881-019-0772-4