
Abstract
Background
Congenital NBCe1A deficiency with the SLC4A4 mutation causes severe proximal renal tubular acidosis, which often comprises extrarenal symptoms, such as intellectual disability and developmental delay, glaucoma, cataract and band keratopathy. To date, almost all mutations have been found to be homozygous mutations located in exons.
Case presentation
We performed direct nucleotide sequencing analysis of exons and exon–intron boundary regions of the SLC4A4 in a patient presenting with severe renal proximal tubule acidosis, glaucoma and intellectual disability and her parents without these signs. The examination revealed compound heterozygous mutations in exon–intron boundary regions, c.1076 + 3A > C and c.1772 − 2A > T, neither of which have been reported previously. While the former mutation was found in the mother, the latter was found in the father. The transcript of the SLC4A4 gene was almost undetectable, and the patient was also diagnosed with Turner’s syndrome.
Conclusions
We identified two novel SLC4A4 mutations, c.1076 + 3A > C and c.1772 − 2A > T. When presented in a compound heterozygous state, these mutations caused a phenotype of severe renal proximal tubular acidosis along with glaucoma and mental retardation. This is the first report of congenital proximal renal tubular acidosis carrying compound heterozygous SLC4A4 mutations in exon–intron boundary regions. We suggest that an mRNA surveillance mechanism, nonsense-mediated RNA decay, following aberrant splicing was the reason that the SLC4A4 transcript was almost undetectable in the proband.
Similar content being viewed by others

Background
The electrogenic sodium bicarbonate cotransporter type 1 (NBCe1) is involved in sodium-coupled bicarbonate transport and has three variants: NBCe1A, which is mainly expressed in the kidney proximal tubule; NBCe1B, in the pancreatic ducts and salivary gland and NBCe1C, in the brain astrocytes. NBCe1A plays an essential role in the regulation of acid–base homeostasis by reabsorbing sodium and bicarbonate filtered in the glomeruli. NBCe1A is encoded by a member of the solute carrier family SLC4A4. Mutations in SLC4A4 cause severe proximal renal tubular acidosis (pRTA), with a plasma pH of 7.04–7.27 and a plasma bicarbonate concentration of 3–17 mmol/L. These patients often present with other extrarenal symptoms, such as glaucoma, band keratopathy and growth retardation [1, 2] (OMIM 604278).
To date, 15 mutations of NBCe1A have been found as follows: 13 homozygous (p.Gln29* [3], p.Arg298Ser [1, 4], p.Ser427Leu [5], p.Thr485Ser [6], p.Gly486Arg [7], p.Arg510His [1], p.Trp516* [8], p.Leu522Pro [9], p.Asp721Thrfs*30 [10], p.Leu738del [11], p.Ala799Val [6], p.Arg881Cys [6] and p.Ser982Aspfs*4 [12]), one compound-heterozygous, p.Arg510His and p.Gln913Arg [13], and one 3’-UTR mutation creating an AU-rich element, c.*206G > A [14]. All these mutations exhibit moderate–to–severe pRTA, except c.*206G > A. Moreover, these mutations are located in the coding region, except p.Ser982Aspfs*4, which involves the intron and generates a premature stop codon and c.*206G > A. To date, no symptomatic mutations with the involvement of splice sites have been found for SLC4A4.
Here, we report a case of compound heterozygous mutations located in the splicing regions with severe pRTA.
Case presentation
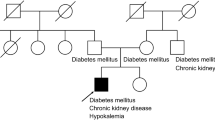
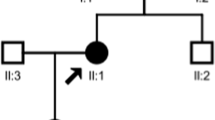
The patient was a 7-year-old Turkish girl born to non-consanguineous parents. She was being followed up since 3 years of age because of bilateral glaucoma and was prescribed medicines [50 mL of Sholl solution and anti-acidosis capsule (three times a day)] and eye drops (β-blocker and carbonic anhydrase inhibitor). Her mother had oligohydramnios; the patient had intrauterine growth retardation and was born prematurely. There was no family history of any inherited diseases, cataract or pRTA. Both her weight and height were below the 3rd percentile, and she demonstrated intellectual disability. However, the other physical examinations, including neurological signs, were unremarkable.
The laboratory tests were as follows: Na, 139 mmol/L; K, 3.1 mmol/L; Cl, 110 mmol/L; blood urea nitrogen, 12 mg/dL and creatinine, 0.9 mg/dL. Blood gas analysis revealed a pH of 7.22, HCO3− concentration of 11 mmol/L and PCO2 of 29 mmHg. In addition, urinalysis revealed a pH of 5, no protein and no glucose. The urinary excretion of amino acids was normal, and the urinary β2-microglobulin level was 110 μg/L (normal: < 240 μg/L). These investigations revealed that the patient had pRTA without Fanconi syndrome – generalized dysfunction of proximal tubule. Considering her short stature, the levels of thyroid hormones, IGF-I and IGFBP3 were normal. Furthermore, whereas the renal ultrasound revealed a 9-mm diameter parenchymal stone in the right kidney, brain MR imaging revealed no intracranial calcification. Neither of the parent showed these symptoms.
At the age of 9, our patient presented with micrognathia, fish-mouth, epicanthal folds, ptosis, low-set ears, a short neck with a low hairline, a broad shield-like chest, wide-spaced nipples, hypoplastic areolae, cubitus valgus and short fourth metacarpals, with other symptoms due to NBCe1A absence such as dental abnormalities, suggesting the coincidence of Turner’s syndrome. In addition, her weight and height were still below the 3rd percentile. Hormonal investigation data were as follows: FSH, 69.3 mIU/mL (normal: 4.5–20.0 mIU/mL); LH, 15.9 mIU/mL (3.5–14.0 mIU/mL) and oestradiol, < 5.0 pg/mL, suggesting hypergonadotropic hypogonadism. Her karyotype was 45, XO which confirmed the diagnosis of Turner’s syndrome.
Genetic analysis
From 200 μL of peripheral blood samples obtained from the patient and her parents, we extracted DNA using the QIAamp DNA Blood Mini Kit (Qiagen Inc.) according to the manufacturer’s instructions. Similarly, we extracted RNA from 1 to 2 mL of the peripheral blood sample using the Isogen (Nippon Gene) or the QIAamp RNA Blood Mini Kit (Qiagen Inc.) according to the manufacturer’s instructions. Then, the complementary DNA (cDNA) of the patient was synthesised from the polyA(+) RNA of the peripheral white blood cells using the cDNA Synthesis Kit (Takara) as previously described [15] or the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific) according to the manufacturer’s instructions.
The polymerase chain reaction (PCR) condition used was as follows: denaturation for 9 min at 95 °C, followed by 35 cycles of 95 °C for 1 min, 60 °C for 1 min and 72 °C for 1 min, with a final extension at 72 °C for 7 min. PCRs were performed using a thermal cycler PerkinElmer GeneAmp PCR System 2400 (PerkinElmer Japan, Applied Biosystems Division, Tokyo, Japan). The DNA sequence of each PCR product was determined using the Sanger sequencing method, with the primers shown in Table 1, in an ABI3100 sequence analyser (Life Technologies, Carlsbad, CA). In addition, AmpliTaq™ (Roche) and attached buffers were used for PCR. The primers in Table 1 were used for the analyses of exons and splicing site sequences of SLC4A4.
The sequences of primers used for the detection of β-actin and fragments of SLC4A4 coding sequences were as follows: hACTB748F, 5’-ATTGGCAATGAGCGGTTC-3′, and hACTB979R, 5’-TCTTCATTGTGCTGGGTGC-3′; exon2-3bridgeF, 5’-GTTGGTGGAGATGATTGTTGAC-3′, and exon6-7bridgeR, 5’-GTCATGGAACACCTCATCAGAC-3′; exon5-6bridgeF, 5’-TGCCCACAAGGTTCTTGTTC-3′, and exon8-9bridgeR, 5’-ACCACAGAACCGTCCAGTTC-3′.
The quantitative RT-PCR (qRT-PCR) was performed according to its instructional manual, with TaqMan Gene Expression Master Mix (Applied Biosystems, Foster City, CA, USA), TaqMan Gene Expression Assays (Hs00186798_m1 for SLC4A4, Hs01060665_g1 for β-actin; all from Applied Biosystems) and sequence detection system (7500 Fast Real-time PCR System; Applied Biosystems). The expression level was quantified relative to the abundance of β-actin cDNA.
Identification of SLC4A4 gene mutations
The sequencing analysis of the SLC4A4 gene (OMIM 603345, ENST00000340595.3, NM_003759.3) across each exon, including the adjacent intronic sequences of approximately 100 base pairs of the proband, revealed two heterozygous mutations as follows: (a) c.1076 + 3A > C, three bases after the end of exon 7 (Fig. 1a and b) c.1772 − 2A > T, two bases before the beginning of exon 12 (Fig. 1b). In addition, we analysed the SLC4A4 genes of her parents and confirmed that her mother and father had heterozygous mutations c.1076 + 3A > and c.1772 − 2A > T, respectively. No other mutations in the SLC4A4 gene were detected in the genomes of the patient or her parents. Of note, both mutations are absent from the ExAC database (http://exac.broadinstitute.org/).

Identification of two novel SLC4A4 mutations. The sequence analysis for the proband and the parents revealed the presence of compound heterozygous mutations c.1076 + 3A > C (a) and c.1772 − 2A > T (b)
In silico analysis
Owing to the locations of both mutations on the splice sites, we performed in silico assays to elucidate whether the splicing sites were altered in the proband.
We used the webtools ‘Splice Site Score Calculation’ (http://rulai.cshl.edu/new_alt_exon_db2/HTML/score.html) [16, 17], ‘NetGene2 Server’ (http://www.cbs.dtu.dk/services/NetGene2/), ‘Human Splicing Finder Version 3.1’ (http://www.umd.be/HSF3/index.html) and ‘Berkeley Drosophila Genome Project Splice Site Prediction by Neural Network’ (http://www.fruitfly.org/seq_tools/splice.html) for the in silico evaluation of these mutations. The ‘Splice Site Score Calculation’ demonstrated that the scores of the original sequences were 9.2 and 9.8, whereas the scores of the aberrant sequences were 2.5 and − 1.2, respectively (in order of c.1076 + 3A > C, c.1772 − 2A > T). Because the mean score of the 3′ splice site in constitutive exons was 7.9 and that of the 5′ splice site in constitutive exons was 8.1, the proband’s data suggested that the mutations could cause aberrant splicing (data not shown).
In contrast, ‘NetGene 2 Server’ suggested that there may be no splice donor site for the c.1076 + 3A > C mutation and that there may be an aberrant acceptor splice site in c.1772 − 2A > T (data not shown), whereas The ‘Human Splicing Finder Version 3.1’ suggested that in c.1772 − 2A > T the acceptor splice site is broken (data not shown). The ‘Berkeley Drosophila Genome Project Splice Site Prediction by Neural Network’ [18] suggested that c.1076 + 3A > C mutation abolishes the original splice donor site and provides an alternative splice donor site (c.1076 + 197_198GT). It also suggested that c.1772 − 2A > T abolishes the original acceptor site and provides alternative acceptor sites (c.1772 − 29_ − 28AG, c.1772 − 37_ − 36AG, c.1772 − 168_ − 167AG).
Furthermore, the estimated models of aberrant transcription according to previous literature [19, 20] suggested the appearance of nonsense codons in each allele of the patient’s genome (Fig. 2a and b).

Models of aberrant transcription caused by splice site mutations. Models of exon skipping by c.1076 + 3A > C mutation (a) and c.1772-2A > T mutation (b) are shown. Alternatively, models of intron retention and activation of cryptic splice sites by c.1076 + 3A > C mutation (c) and c.1772-2A > T mutation (d) are shown. Cryptic splice sites are predicted using Splice Site Prediction by Neural Network in Berkeley Drosophila Genome Project. All the four models predict premature stop codons
The analysis of the patient’s cDNA
We tried to assess the sequence of the SLC4A4 cDNA because we obtained the cDNA of the proband from her mRNA. However, we did not detect the expression of SLC4A4 (Fig. 3a). Then, owing to the detection of the expression of β-actin in the proband’s cDNA (Fig. 3b), the absence of the SLC4A4 cDNA sequence suggested that SLC4A4 was either not expressed or expressed at extremely low levels in the proband.

Identification of fragments of SLC4A4 coding sequences (a) and β-actin (as a control; b) in cDNA of the proband and control healthy human. Reverse-transcription PCR (RT-PCR) products of the SLC4A4 coding region were not detected in the proband. In contrast, the fragment of β-actin was detected in the proband’s cDNA. PT, proband; P/C, positive control (the cDNA from white blood cells of a healthy human as a template); N/C, negative control (no template). c quantitative RT-PCR (qRT-PCR) in cDNA provided by the proband, parents and control. n = 2 each. ***: p < 0.01, **: P < 0.05, *: P < 0.1
We further tried to confirm the expression amount of the SLC4A4 compared to β-actin in the patient by the qRT-PCR. The relative expression ratio of SLC4A4 to β-actin in the proband was extremely low compared to that of the healthy human control (P < 0.01), mother (P < 0.05) and father (P < 0.1) (Fig. 3c).
Discussion and conclusions
To the best of our knowledge, this is the first report of pRTA caused by SLC4A4 mutation with unique aberrant sequences and compound heterozygous mutations that are both splice site mutations.
Typical patients with NBCe1A deficiency present with severe pRTA accompanied by extrarenal symptoms, such as ocular symptoms – band keratopathy, cataract and glaucoma, growth and mental retardation and sometimes intracranial calcification [4]. This patient also had severe pRTA (HCO3− concentration: 11 mmol/L), glaucoma and growth and mental retardation, significantly coinciding with typical pRTA phenotypes because of NBCe1A deficiency.
All cases of previously determined pRTA because of the NBCe1A mutation had mutations involving exons; some mutations showed intracellular retention (p.Arg298Ser, p.Arg510His, p.Arg881Cys and p.982Serfs*4) or no protein expression (p.Gln29* and p.Trp516*) in polarised MDCK cells. Functional analyses of p.Arg298Ser, p.Thr485Ser, p.Gly486Arg, p.Arg510His, p.Leu522Pro, p.Asn721Thrfs*30, p.Ala799Val, p.Arg881Cys and p.982Serfs*4 elucidated the pathophysiology of NBCe1A deficiency, the acid–base regulation of the proximal tubule and the functional roles of NBCe1A [6, 7, 10, 12, 21]. When expressed in Xenopus oocytes or HEK293 cells, the function of this mutant NBCe1A was reduced (p.Arg298Ser, p.Thr485Ser, p.Gly486Arg, p.Arg510His and p.Arg881Cys) or almost abolished (p.Leu522Pro, p.Asn721Thrfs*30 and p.Ala799Val), as determined by electrophysiological analysis and/or cell pH measurement.
In our patient, reverse-transcription PCR (RT-PCR) revealed that SLC4A4 mRNA was not detected in the peripheral leukocytes, although the primers were designed well upstream of the mutations. Moreover, qRT-PCR showed that the expression ratio of SLC4A4 to β-actin was extremely low in the patient. Hence, we could not identify the exact sequence of the aberrantly spliced SLC4A4 mRNA in the patient. Because we could detect the expression of the house-keeping gene, β-actin in the patient’s cDNA, the most probable speculation would be that the expression level of SLC4A4 mRNA was at extremely low levels or almost zero in this patient.
Perhaps, the extremely low expression of SLC4A4 in the qRT-PCR and abundance below detection limit of SLC4A4 mRNA in the RT-PCR could be attributed to some type of mRNA surveillance, a mechanism that controls the quality of mRNA in eukaryotes [22, 23]. Three types of mRNA surveillance found are nonsense-mediated mRNA decay (NMD), nonstop decay (NSD) and no-go decay (NGD) [24,25,26,27]. NMD was first identified in mRNA surveillance and is widely known to accelerate the degradation of aberrant mRNAs having a premature terminate codon. While NSD is a mechanism that degrades mRNAs lacking a stop codon, NGD eliminates mRNAs with elongation stalls. Mutations in the borderline splicing regions may cause exon skipping, the activation of cryptic splice sites and the creation of a pseudo-exon within an intron and/or intron retention [28].
Figure 2a and b show the estimated models of exon skipping, because previous literatures indicated that mutations in splice donor sites [19, 29, 30] and splice acceptor sites [31, 32] can cause exon skipping. Figure 2a shows a model that an A > C change at position + 3 of the splice donor site of intron 7 causes skipping of exon 7, which results in a premature stop codon. Similarly, Fig. 2b shows a model that an A > T change at position − 2 of the splice acceptor site upstream of exon 12 causes skipping of exon 12, which results in a premature stop codon [20, 31, 32].
Other possible consequences of the splice site mutations would be intron retention and activation of cryptic splice sites. The all cryptic splice sites suggested by ‘Berkeley Drosophila Genome Project Splice Site Prediction by Neural Network’, a splice donor site (c.1076 + 197_198 corresponding to GU at the 5′ end of the intron) generated by c.1076 + 3A > C, and splice acceptor sites (c.1772 − 29_ − 28, c.1772 − 37_ − 36, and c.1772 − 168_ − 167 corresponding to AG at the 3′ end of the intron) generated by c.1772 − 2A > T, are predicted to cause premature stop codons (Fig. 2c and d) when activated. These aberrantly spliced mRNAs through exon skipping or activation of cryptic splice sites may have been degraded by NMD because of premature stop codons.
Hence, we speculate the occurrence of NMD in our patient, resulting in the absence of NBCe1A and the pathogenesis of pRTA. Because SLC4A4 cDNAs were detected in the peripheral leukocytes of patients with most of previously reported mutations [1, 3, 5,6,7,8,9,10,11,12,13, 21], this study revealed a novel mechanism in congenital NBCe1A deficiency.
Turner’s syndrome is one of the most common genetic disorders, resulting from the complete or partial deletion of one X chromosome [33, 34]. The clinical features of Turner’s syndrome usually comprise symptoms such as short stature, abnormalities as mentioned earlier, ovarian insufficiency, and cardiac complications. While the reason for the delayed diagnosis of Turner’s syndrome remains unclear, this delay occurs very often, as mentioned in previous studies [33]. In our patient, the growth retardation was first attributed to congenital pRTA, which may have delayed the diagnosis of Turner’s syndrome. The total clinical timeline is summarized in Table 2.
In conclusion, we identified novel compound heterozygous splice site mutations of SLC4A4 in a patient presenting typical pRTA. In our opinion, NBCe1A deficiency caused by NMD, a type of mRNA surveillance mechanisms, could be held accountable for these symptoms.
Abbreviations
- NMD:
-
Nonsense-mediated decay
- PCR:
-
Polymerase chain reaction
- pRTA:
-
Proximal renal tubular acidosis
- cDNA:
-
Complementary DNA
- NSD:
-
Nonstop decay
- NGD:
-
No-go decay
References
Igarashi T, Inatomi J, Sekine T, Cha SH, Kanai Y, Kunimi M, Tsukamoto K, Satoh H, Shimadzu M, Tozawa F, et al. Mutations in SLC4A4 cause permanent isolated proximal renal tubular acidosis with ocular abnormalities. Nat Genet. 1999;23(3):264–6.
Seki G, Horita S, Suzuki M, Yamazaki O, Usui T, Nakamura M, Yamada H. Molecular mechanisms of renal and extrarenal manifestations caused by inactivation of the electrogenic Na(+)-HCO3 (−) cotransporter NBCe1. Front Physiol. 2013;4:270.
Igarashi T, Inatomi J, Sekine T, Seki G, Shimadzu M, Tozawa F, Takeshima Y, Takumi T, Takahashi T, Yoshikawa N, et al. Novel nonsense mutation in the Na+/HCO3- cotransporter gene (SLC4A4) in a patient with permanent isolated proximal renal tubular acidosis and bilateral glaucoma. J Am Soc Nephrol. 2001;12(4):713–8.
Igarashi T, Sekine T, Inatomi J, Seki G. Unraveling the molecular pathogenesis of isolated proximal renal tubular acidosis. J Am Soc Nephrol. 2002;13(8):2171–7.
Dinour D, Chang MH, Satoh J, Smith BL, Angle N, Knecht A, Serban I, Holtzman EJ, Romero MF. A novel missense mutation in the sodium bicarbonate cotransporter (NBCe1/SLC4A4) causes proximal tubular acidosis and glaucoma through ion transport defects. J Biol Chem. 2004;279(50):52238–46.
Horita S, Yamada H, Inatomi J, Moriyama N, Sekine T, Igarashi T, Endo Y, Dasouki M, Ekim M, Al-Gazali L, et al. Functional analysis of NBC1 mutants associated with proximal renal tubular acidosis and ocular abnormalities. J Am Soc Nephrol. 2005;16(8):2270–8.
Suzuki M, Vaisbich MH, Yamada H, Horita S, Li Y, Sekine T, Moriyama N, Igarashi T, Endo Y, Cardoso TP, et al. Functional analysis of a novel missense NBC1 mutation and of other mutations causing proximal renal tubular acidosis. Pflugers Arch. 2008;455(4):583–93.
Lo YF, Yang SS, Seki G, Yamada H, Horita S, Yamazaki O, Fujita T, Usui T, Tsai JD, Yu IS, et al. Severe metabolic acidosis causes early lethality in NBC1 W516X knock-in mice as a model of human isolated proximal renal tubular acidosis. Kidney Int. 2011;79(7):730–41.
Demirci FY, Chang MH, Mah TS, Romero MF, Gorin MB. Proximal renal tubular acidosis and ocular pathology: a novel missense mutation in the gene (SLC4A4) for sodium bicarbonate cotransporter protein (NBCe1). Mol Vis. 2006;12:324–30.
Inatomi J, Horita S, Braverman N, Sekine T, Yamada H, Suzuki Y, Kawahara K, Moriyama N, Kudo A, Kawakami H, et al. Mutational and functional analysis of SLC4A4 in a patient with proximal renal tubular acidosis. Pflugers Arch. 2004;448(4):438–44.
Kari JA, El Desoky SM, Singh AK, Gari MA, Kleta R, Bockenhauer D. The case | renal tubular acidosis and eye findings. Kidney Int. 2014;86(1):217–8.
Suzuki M, Van Paesschen W, Stalmans I, Horita S, Yamada H, Bergmans BA, Legius E, Riant F, De Jonghe P, Li Y, et al. Defective membrane expression of the Na(+)-HCO(3)(−) cotransporter NBCe1 is associated with familial migraine. Proc Natl Acad Sci U S A. 2010;107(36):15963–8.
Myers EJ, Yuan L, Felmlee MA, Lin YY, Jiang Y, Pei Y, Wang O, Li M, Xing XP, Marshall A, et al. A novel mutant Na+ /HCO3- cotransporter NBCe1 in a case of compound-heterozygous inheritance of proximal renal tubular acidosis. J Physiol. 2016;594(21):6267–86.
Patel N, Khan AO, Al-Saif M, Moghrabi WN, AlMaarik BM, Ibrahim N, Abdulwahab F, Hashem M, Alshidi T, Alobeid E, et al. A novel mechanism for variable phenotypic expressivity in Mendelian diseases uncovered by an AU-rich element (ARE)-creating mutation. Genome Biol. 2017;18(1):144.
Nakamura M, Satoh N, Suzuki M, Kume H, Homma Y, Seki G, Horita S. Stimulatory effect of insulin on renal proximal tubule sodium transport is preserved in type 2 diabetes with nephropathy. Biochem Biophys Res Commun. 2015;461(1):154–8.
Stamm S, Zhu J, Nakai K, Stoilov P, Stoss O, Zhang MQ. An alternative-exon database and its statistical analysis. DNA Cell Biol. 2000;19(12):739–56.
Liu HX, Cartegni L, Zhang MQ, Krainer AR. A mechanism for exon skipping caused by nonsense or missense mutations in BRCA1 and other genes. Nat Genet. 2001;27(1):55–8.
Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in genie. J Comput Biol. 1997;4(3):311–23.
Koppolu AA, Madej-Pilarczyk A, Rydzanicz M, Kosinska J, Gasperowicz P, Dorszewska J, Kozubski W, Steinborn B, Kochanski AM, Ploski R. A novel de novo COL6A1 mutation emphasizes the role of intron 14 donor splice site defects as a cause of moderate-progressive form of ColVI myopathy - a case report and review of the genotype-phenotype correlation. Folia Neuropathol. 2017;55(3):214–20.
Zavodna K, Bujalkova M, Krivulcik T, Alemayehu A, Skorvaga M, Marra G, Fridrichova I, Jiricny J, Bartosova Z. Novel and recurrent germline alterations in the MLH1 and MSH2 genes identified in hereditary nonpolyposis colorectal cancer patients in Slovakia. Neoplasma. 2006;53(4):269–76.
Yamazaki O, Yamada H, Suzuki M, Horita S, Shirai A, Nakamura M, Satoh N, Fujita T, Seki G. Identification of dominant negative effect of L522P mutation in the electrogenic Na(+)-HCO(3)(−) cotransporter NBCe1. Pflugers Arch. 2013;465(9):1281–91.
Shoemaker CJ, Green R. Translation drives mRNA quality control. Nat Struct Mol Biol. 2012;19(6):594–601.
Kurosaki T, Maquat LE. Nonsense-mediated mRNA decay in humans at a glance. J Cell Sci. 2016;129(3):461–7.
Doma MK, Parker R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nat. 2006;440(7083):561–4.
Tollervey D. Molecular biology: RNA lost in translation. Nat. 2006;440(7083):425–6.
Colobran R, Lois S, de la Cruz X, Pujol-Borrell R, Hernandez-Gonzalez M, Guilarte M. Identification and characterization of a novel splice site mutation in the SERPING1 gene in a family with hereditary angioedema. Clin Immunol. 2014;150(2):143–8.
Harigaya Y, Parker R. No-go decay: a quality control mechanism for RNA in translation. Wiley Interdiscip Rev RNA. 2010;1(1):132–41.
Nakai K, Sakamoto H. Construction of a novel database containing aberrant splicing mutations of mammalian genes. Gene. 1994;141(2):171–7.
Souma T, Tompson SW, Thomson BR, Siggs OM, Kizhatil K, Yamaguchi S, Feng L, Limviphuvadh V, Whisenhunt KN, Maurer-Stroh S, et al. Angiopoietin receptor TEK mutations underlie primary congenital glaucoma with variable expressivity. J Clin Invest. 2016;126(7):2575–87.
Jackson MZ, Gruner KA, Qin C, Tourtellotte WG. A neuron autonomous role for the familial dysautonomia gene ELP1 in sympathetic and sensory target tissue innervation. Dev. 2014;141(12):2452–61.
Sasai H, Aoyama Y, Otsuka H, Abdelkreem E, Nakama M, Hori T, Ohnishi H, Turner L, Fukao T. Single-nucleotide substitution T to a in the polypyrimidine stretch at the splice acceptor site of intron 9 causes exon 10 skipping in the. Mol Genet Genomic Med. 2017;5(2):177–84.
Silveira LF, Stewart PM, Thomas M, Clark DA, Bouloux PM, MacColl GS. Novel homozygous splice acceptor site GnRH receptor (GnRHR) mutation: human GnRHR “knockout”. J Clin Endocrinol Metab. 2002;87(6):2973–7.
Lee MC, Conway GS. Turner’s syndrome: challenges of late diagnosis. Lancet Diabetes Endocrinol. 2014;2(4):333–8.
Stochholm K, Juul S, Juel K, Naeraa RW, Gravholt CH. Prevalence, incidence, diagnostic delay, and mortality in turner syndrome. J Clin Endocrinol Metab. 2006;91(10):3897–902.
Funding
SH, HI, MoN, and NS received JSPS KAKENHI Grant Number 16 K09640, 17H05085. 17 K16071, and 16H06750 respectively. ST received Grant Number 16kk0205001h0001 from AMED.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files, Table 1, Table 2, Fig. 1a/b, Fig. 2a/b and Fig. 3a/b.
Author information
Authors and Affiliations
Contributions
SH, HI and GS designed and organized the study. ES, TS and NY sampled the family members and acquired the clinical data. SH, ES, HI, MoN, NS, AS HT and TM carried out the molecular genetic studies. SH, ES, TS, NY, HI, MoN, NS, AS, HT, TM, GS, and ST analyzed and interpreted the molecular and the clinical data. SH wrote the manuscript that was edited by ES, HI, GS, ST and MaN. All authors have read and approved the final version of the manuscript submitted by SH.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
We obtained written informed consent for genetic analysis from the patient and her parents, in compliance with the Declaration of Helsinki. This consent was approved by the ethical committees of the University of Tokyo Faculty of Medicine and Eskisehir Osmangazi University School of Medicine.
Consent for publication
We obtained written informed consent for the publication of medical data of their child from her parents, in compliance with the Declaration of Helsinki.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Horita, S., Simsek, E., Simsek, T. et al. SLC4A4 compound heterozygous mutations in exon–intron boundary regions presenting with severe proximal renal tubular acidosis and extrarenal symptoms coexisting with Turner’s syndrome: a case report. BMC Med Genet 19, 103 (2018). https://doi.org/10.1186/s12881-018-0612-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-018-0612-y