
Abstract
Background
FGFR2 encodes a fibroblast growth factor receptor whose mutations are responsible for the Crouzon syndrome, involving craniosynostosis and facial dysostosis with shallow orbits. However, few reports are available quantifying the orbital volume of Crouzon syndrome and there was little direct evidence to show FGFR2 mutation actually influencing orbital morphology.
Methods
Ten Crouzon syndrome patients underwent a standard ophthalmologic assessment. Morphology study was carried out based on 3-dimensional computed tomography scan to calculate orbital volume. Genomic DNA was extracted from peripheral blood leukocytes of the patients and genomic screening of FGFR2. A three-dimensional computer model was used to analyse the structural positioning of the mutation site that was predicted possible impact on functional of FGFR2 protein. Real-time PCR was performed to analyse the expression of bone maker gene.
Results
We describe a FGFR2 mutation (p.G338R, c.1012G > C) in a Chinese family with Crouzon syndrome. Computational analysis showed the mutate protein obviously changes in the local spatial structure compared with wild-type FGFR2. The expression of osteocalcin and alkaline phosphatase two osteoblast specific genes significantly increased in orbital bone directly from patient compared to normal individual, which may lead to facial dysostosis. This is compatible with the shallow and round orbits in our Crouzon syndrome patient.
Conclusions
Our study further identified G338R FGFR2 mutation (c1012G > C) lead to inherited Crouzon syndrome. Thus, early intervention, both medically and surgically, as well as disciplined by a multiple interdisciplinary teams are crucial to the management of this disorder.
Similar content being viewed by others

Background
Crouzon syndrome is characterized by craniosynostosis and facial dysostosis with an incidence of 16.5/1,000,000, accounting for 4.8% of all craniosynostoses [1]. In ophthalmology, ocular involvement can include variations ranging from exophthalmos and divergent strabismus to ocular hypertelorism [2]. The proptosis which can in turn put patients at increased for conjunctivitis, is secondary to shallow orbits. Currently, Crouzon syndrome is commonly diagnosed when patients present with obviously facial structure abnormalities and growth patterns. However, exophthalmos was happening largely before these presentations as exophthalmos was visualized on fetal ultrasound at 35 weeks for orbital growth retardation [3]. Unfortunately, few reports are available quantifying the orbital volume of Crouzon syndrome.
Molecular findings prove it is a rare autosomal dominant disease. Mutations in FGFR2, a gene encoding fibroblast growth factor receptor type 2 (FGFR2) located at 10q26 have been identified in both syndromic and non-syndromiccraniosynostosis [4,5,6,7]. It consists of three extracellular immunoglobulainlike (Ig) domains, a single transmembrane segment, and a split tyrosine kinase domain [8]. Although a number of mutations of FGFR2 have been well characterized to date and a few mutations are identified affecting cell proliferation such as osteoblasts and chondrocytes in vitro, [9,10,11] however, to our knowledge, there is no literature reported the expression of osteoblasts and chondrocytes in vivo, especially in orbital bone from Crouzon syndrome patient.
In this manuscript, we have attempted to identify the gain-of-function role of G338R FGFR2 (c.1012G > C) potentially related to shallow orbits even premature suture closure in Crouzon syndrome patient. We found OC (osteocalcin) and ALP (alkaline phosphatase) two selected bone marker genes were significantly increased in the patient’s orbital bone which indicate G338R FGFR2 may affect orbital bone development.
Methods
Ethics statement and clinic examination
The research was carried out according to the principles of the Declaration of Helsinki; informed consent was obtained and Shanghai Ninth People’s Hospital Ethics Committee approved the study. The patient photos, which are contained in this article, were taken by a hospital-based photographer at Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine. Permission to use these photos in this report has been obtained from all the subjects including the parents of individual V2 and IV-2 (minor’s parent: legal guardian) who participated in this study.
We present 10 Crouzon syndrome patients from one Chinese family with 5 generation. All patients underwent a standard ophthalmologic assessment. Three-dimensional orbital computed tomography (CT) scan examination was used for bony defects. All CT scans were obtained with a spiral CT scanner (GE LightSpeed 16, Milwaukee, WI). The scans included the range from the maxilla to the skull. Continuous high-resolution scanning with a slice detection of 1.25*16 mm, reconstructed thickness of 1.25 mm, and slice increment of 1.25 mm was performed. The reconstructed field of view was 23 cm, and the reconstruction matrix was 512*512. All CT images were loaded in the DICOM format. Mimics v18.0 software (Materialise, Leuven, Belgium) was used for further calculations and analyses. After the DICOM data were imported into Mimics, the coordinate system was redefined to ensure that all measurements were completed in the same environment. The new coordinate system was defined according to the protocols of the “Online Reslice” and “Reslice Project” functions. The axial plane was defined as a plane parallel to the Frankfurt plane, passing through the inferior margin of the left orbit and the upper margin of each of the external auditory meatuses. The sagittal plane was defined as the median plane passing through the midpoint of the nasion, sellaturcica, and foramen magnum. The oblique sagittal plane was defined as the plane passing through the midpoint of the optic canal orifice and the ipsilateral orbital aperture and was perpendicular to the axial plane. The coronal datum plane was defined as the coronal plane passing through the most posterior point of the lateral orbital rim. The orbital width, orbital height, roof length, floor length, medial wall length, lateral wall length, ocular protrusion, bony orbital volume (BOV), midinterorbital distance and lateral orbital wall angle parameters were calculated according to the protocol in the previous study [12].
FGFR2 mutation screening
Genomic DNA was extracted from peripheral blood leukocytes of the patients and 100 randomly selected healthy volunteers using the Automatic Nucleic Acid Isolation System (QuickGene-610 L; Fujifilm Life Science, Tokyo, Japan). PCR for the target sequence of FGFR2 was performed, the products purified and sequenced as previously described [13, 14]. A three dimensional computer model was used to analyse the structural positioning of FGFR2-G338A mutation site. The immunoglobulin (Ig)-like domain 3 of FGFR2 protein was modeled with SWISS-MODEL (template PDB code, 1iil) as we have previously described [15, 16].
Real-time00205PCR for osteocalcin (OC) and alkaline phosphatase (ALP)
Orbital bones obtained from Crouzon syndrome patient and orbital fracture patient on patient’s consent. After impurities removing, bone samples were soaked in lquid nitrogen and grined into fine particles or a powder. Total RNA was extracted using TRI-REAGENT (Invitro- gen, USA) according to the manufacturer’s instructions, and cDNA was synthesized using the PrimeScript RT re-agent kit (Takara, Japan) as we have previously described [17]. The housekeeping gene GAPDH was used as an endogenous control. The PCR cycle parameters for OC and ALP expression were as follows: 33 cycles of denaturation at 95 °C for 30 s, optimal annealing temperature for 30 s, extension at 72°Cfor30sandafinalextensionat72°Cfor5min.
All of the experiments were performed in triplicate, and the data were expressed as the mean ± standard deviation (SD). The comparative threshold cycle method was applied in the quantitative real-time RT-PCR assay according to the ΔΔ threshold cycle method.
Results
Patients and clinical characteristics
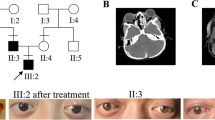
We present 10 Crouzon syndrome patients from one Chinese family with 5 generation (Fig. 1a). These patients had shallow orbits and ocular proptosis, accompanied by craniosynostosis, mid-face hypoplasia, a curved, beaklike nose, but clinically normal hands and feet. They had normal vision since early childhood, just displaying a surprised look, but their vision was getting worse and worse as they aged. The visual acuity of individual III-14 (unmarried, 64 years old) was FC /1 m (figure count, OU). The quality of his life is at stake for serious exposure keratitis and obstinate conjunctivitis. And even more severe visual impairment, because of chronic corneal scarring result from hypophasis (Fig. 1b, III21and IV16), the visual acuity of III21 and IV16 was LP (light perception, OU of III21, OS of IV16).

Crouzon syndrome patients and morphology measurement of orbit. a Five-generation Crouzon syndrome pedigree. Affected individuals are indicated by filled symbols, the proband is marked with an upward arrow. b Facial photographs of the Crouzon syndrome patients. c Morphology measurement of orbit. (Ca) Three-dimensional reconstruction of skull of the proband IV-2 (Cb) Morphology measurement of orbit(a. midinterorbital distance, b.lateral orbital wall angle) (Cc) Morphology measurement of orbit(a. roof length, b. medial wall length, c. floor length, d. lateral wall length, e. orbital width, f. orbital height)
Individual IV-2 (Fig. 1b, panel 1) the probands was born after a normal pregnancy and was referred for craniofacial assessment at 14 years of age because prominent eyes. He had a Crouzon syndrome brother (IV-1) died around 8 months old. The visual acuity of the IV-2 patient was 0.4 (OD) and 0.1 (OS). The refractive error was − 2.25D, with − 1.00 astigmatism (OD) and + 0.75D, with − 3.00 astigmatism (OS). Vertical interpalpebral fissure height (IPFH) OD 11 mm, OS 7 mm; horizontal palpebral fissure length (HPFL) OD 20 mm, OS 19 mm; levator function OD 5 mm, OS 8 mm; The prominence of the IV-2 patient was 20 mm (OD) and 18 mm (OS). In addition aural atresia was detected. No abnormalities were detected in the lenses, retinas, choroids, or optic nerves.
Orbital morphology
To measurement the orbital volume, we carried out the morphology study. The data showed the orbital volume of Crouzon syndrome patient was much more shallower and rounder than normal population (Table 1). As the data show the shape of the orbital aperture was more round and bigger than the normal orbits, for both the orbital height and width were larger than what we have previously measured from normal male Chinese adults [12] for the bony orbit have almost reached the adult level in the 14-year old patient [18]. The patient orbits were quite shallow as all length parameters from the orbital aperture to the optic nerve canal orifice were smaller than the normal data, especially the medial wall length (OD 39.97 mm, OS 38.00 mm, normal 46.32 ± 2.67 mm) and the floor length (OD 37.38 mm, OS 34.66 mm, normal 47.93 ± 2.68 mm), which was consistent with the dysplasia of the maxilla,finally resulted in a small orbital cavity (OD 17.15 ml, OS 16.31 ml, normal 26.04 ± 2.60 ml).However,the patient ocular volume was 5.76 ml (OD) and 5.96 ml (OS)which was normal along with the shallow orbits finally caused the “uninhabitable habitation”, then exophthalmos was presented.
G338R mutation was identified in FGFR2 (Ig)-like domain
Sequencing of the complete coding sequence of the FGFR2 gene derived from genomic DNA revealed a heterozygous mutation (p.G338R, c. 1012G > C) in exon 10 of the human FGFR2 (Fig. 2a), causing the replacement of glycine (Gly) by arginine (Arg) at amino acid position 338, a highly conserved segment of the FGFR2 protein (Fig. 2c). This mutation is located in the immunoglobulin (Ig)-like domain 3 (Fig. 2b). Moreover, the missense mutation was not found in 100 normal people, including III-5 and III-7 from the Crouzon syndrome family who were normal subjects (Fig. 2a).

A heterozygous missense mutation was identified in FGFR2. a A heterozygous missense mutationin exon 10 of the FGFR2 was identified in Crouzon syndromepatients which was absent in normal population. b Mapping of pathogenic FGFR2 mutations. c Amino acid sequence of FGFR2 around G338R in species of the vertebrates and FGFR family, a highly conserved segment of the FGFR2 protein. d Molecular modelling of the immunoglobulin (Ig)-like domain 3 of the wild-type and mutant-typep.G338RFGFR2
To study the structural changes in the mutant-type FGFR2, three-dimensional structural model was used to analyse the position of missense mutation. Compared with wild-type FGFR2 (Fig. 2d, a-c), obviously changes were detected in the local spatial structure of the mutant-type FGFR2 (Fig. 2d, d-f) carrying the amino acid substitution in the (Ig)-like domain 3. Notably, in the wild-type FGFR2 there was no hydrogen bond between Tyr328 and Arg330 (Fig. 2d, c). However, in the mutant-type FGFR2one hydrogen bond was formed between Tyr328 and Arg330 that was near the Gly338 amino acid residue (Fig. 2d, f), which may reduced the stability of protein.
OC and ALP mRNA level was increased in patient orbital bone
To explore the factors that may affect the upgrowth of orbit bone in Crouzon syndrome patients,we examined two osteoblast related genes expressions OC and ALP. The results clearly showed that OC and ALP was remarkably increased in the Crouzon syndrome patient compared with normal orbital bone (Fig. 3a and b). These data further highlight the clinical importance of FGFR2 in craniofacial growth.

Osteoblast specific genes expression levels in patient and normal orbital bone. Both osteocalcin (a) and alkaline phosphatase (b) were highly expressed in patient’ orbital bone. *P < 0.05, compared with the normal
Discussion
Crouzon syndrome, although of variable penetrance is accounting for 4.8% of all craniosynostoses [1]. Currently, Crouzon syndrome is commonly diagnosed when patients present with obviously facial structure abnormalities and growth patterns. However, exophthalmos was happening largely before these presentations as exophthalmos was visualized on fetal ultrasound at 35 weeks for orbital growth retardation [3]. Clinically, exophthalmos can be diagnosed in pregnancy. Furthermore, through chorionic villus biopsy, Schwartz using PCR by targeting of FGFR2 known mutation found within the pregnant mother, successfully demonstrated diagnosis of the pregnancy Crouzon syndrome [19]. A number of mutations of FGFR2 have been well characterized to date and a few mutations are identified affecting cell proliferation such as osteoblasts and chondrocytes in vitro [9,10,11]. Thus, any minor changes of FGFR2 and the spatiotemporal planes can then inevitably lead to primordial bone changes [20].
Given the scant array of case reports in the literature, treatment of Crouzon syndrome borrows from the general craniosynostosis literature in addition to existing case reports. Ultimately, molecular genetic testing should be performed in the context of genetic counseling, and be complemented by multidisciplinary clinical follow-up by a multiple interdisciplinary teams to preventing the process of traumatic repeat surgeries for patients struck with the disease.
In summary, our results provide direct evidence that FGFR2 mutation is associated with shallow orbits in Crouzonsyndrome that confirms the crucial and conserved role of FGFR2 during orbit development. This finding is useful and valuable for genetic counseling and prenatal diagnosis in families with Crouzon syndrome without serious ocular disorders.
Conclusions
Crouzon syndrome is characterized by exophthalmos and divergent strabismus to ocular hypertelorism in ophthalmology mainly because shallow orbits. Our study identified one FGFR2 mutation (p.G338R, c.1012G > C) that increased osteogenesis gene expression from one Chinese family with 5 generation. The weak findings in this study provide little support for mutation of FGFR2 may lead to reduce the volume of orbit in Crouzon syndrome patients. If future evidence supports this finding, it may provide reassurance to multiple interdisciplinary teams concerned about early intervention on this disorder.
Further research could explore how FGFR2 gene can cause shallow orbits in Crouzon syndrome.
Abbreviations
- ALP:
-
Alkaline phosphatase
- Arg:
-
Arginine
- BOV:
-
Bony orbital volume
- CT:
-
Computed tomography
- FC:
-
Figure count
- Gly:
-
Glycine
- HPFL:
-
Horizontal palpebral fissure length ()
- IPFH:
-
Vertical interpalpebral fissure height
- LP:
-
Light perception
- OC:
-
Osteocalcin
References
Cohen MM Jr, Kreiborg S. Birth prevalence studies of the Crouzon syndrome: comparison of direct and indirect methods. Clin Genet. 1992;41(1):12–5.
Horbelt CV. Physical and oral characteristics of Crouzon syndrome, Apert syndrome, and Pierre Robin sequence. Gen Dent. 2008;56(2):132–4.
Menashe Y, Ben Baruch G, Rabinovitch O, Shalev Y, Katzenlson MB, Shalev E. Exophthalmus--prenatal ultrasonic features for diagnosis of Crouzon syndrome. Prenat Diagn. 1989;9(11):805–8.
Rutland P, Pulleyn LJ, Reardon W, Baraitser M, Hayward R, Jones B, Malcolm S, Winter RM, Oldridge M, Slaney SF, et al. Identical mutations in the FGFR2 gene cause both Pfeiffer and Crouzon syndrome phenotypes. Nat Genet. 1995;9(2):173–6.
Li X, Park WJ, Pyeritz RE, Jabs EW. Effect on splicing of a silent FGFR2 mutation in Crouzon syndrome. Nat Genet. 1995;9(3):232–3.
Kan SH, Elanko N, Johnson D, Cornejo-Roldan L, Cook J, Reich EW, Tomkins S, Verloes A, Twigg SR, Rannan-Eliya S, et al. Genomic screening of fibroblast growth-factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis. Am J Hum Genet. 2002;70(2):472–86.
Mansukhani A, Bellosta P, Sahni M, Basilico C. Signaling by fibroblast growth factors (FGF) and fibroblast growth factor receptor 2 (FGFR2)-activating mutations blocks mineralization and induces apoptosis in osteoblasts. J Cell Biol. 2000;149(6):1297–308.
Plotnikov AN, Hubbard SR, Schlessinger J, Mohammadi M. Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell. 2000;101(4):413–24.
Eswarakumar VP, Monsonego-Ornan E, Pines M, Antonopoulou I, Morriss-Kay GM, Lonai P. The IIIc alternative of Fgfr2 is a positive regulator of bone formation. Development. 2002;129(16):3783–93.
Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16(12):1446–65.
Fanganiello RD, Sertie AL, Reis EM, Yeh E, Oliveira NA, Bueno DF, Kerkis I, Alonso N, Cavalheiro S, Matsushita H, et al. Apert p.Ser252Trp mutation in FGFR2 alters osteogenic potential and gene expression of cranial periosteal cells. Mol Med. 2007;13(7–8):422–42.
Ji Y, Qian Z, Dong Y, Zhou H, Fan X. Quantitative morphometry of the orbit in Chinese adults based on a three-dimensional reconstruction method. J Anat. 2010;217(5):501–6.
Fan J, Zhou Y, Huang X, Zhang L, Yao Y, Song X, Chen J, Hu J, Ge S, Song H, et al. The combination of polyalanine expansion mutation and a novel missense substitution in transcription factor FOXL2 leads to different ovarian phenotypes in blepharophimosis-ptosis-epicanthus inversus syndrome (BPES) patients. Hum Reprod. 2012;27(11):3347–57.
Fan JY, Wang YF, Han B, Ji YR, Song HD, Fan XQ. FOXL2 mutations in Chinese families with Blepharophimosis syndrome (BPES). Transl Res. 2011;157(1):48–52.
Fan JY, Han B, Qiao J, Liu BL, Ji YR, Ge SF, Song HD, Fan XQ. Functional study on a novel missense mutation of the transcription factor FOXL2 causes blepharophimosis-ptosis-epicanthus inversus syndrome (BPES). Mutagenesis. 2011;26(2):283–9.
Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22(2):195–201.
Fan J, Xing Y, Wen X, Jia R, Ni H, He J, Ding X, Pan H, Qian G, Ge S, et al. Long non-coding RNA ROR decoys gene-specific histone methylation to promote tumorigenesis. Genome Biol. 2015;16(1):139.
Ji Y, Ye F, Zhou H, Xie Q, Ge S, Fan X. Bony orbital maldevelopment after enucleation. J Anat. 2015;227(5):647–53.
Schwartz M, Kreiborg S, Skovby F. First-trimester prenatal diagnosis of Crouzon syndrome. Prenat Diagn. 1996;16(2):155–8.
Jabs EW, Li X, Scott AF, Meyers G, Chen W, Eccles M, Mao JI, Charnas LR, Jackson CE, Jaye M. Jackson-Weiss and Crouzon syndromes are allelic with mutations in fibroblast growth factor receptor 2. Nat Genet. 1994;8(3):275–9.
Acknowledgements
We would like to extend our gratitude to our patients who participated in this study.
Funding
This work was supported by The National Natural Science Foundation of China (grants 81502481), Foundation of Ninth People’s Hospital Affiliated to Shanghai JiaoTong University School of Medicine (2014A05) and the Science and Technology Commission of Shanghai (17DZ2260100).
Availability of data and materials
The data that support the findings of this study are not publicly available due to the issue of intellectual property but are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
RBJ and XQF designed the study that was performed and analysed by JYF and YWL. JYF made the first draft of the manuscript that has been critically revised by RBJ and XQF. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The research was carried out according to the principles of the Declaration of Helsinki; informed consent was obtained and Shanghai Ninth People’s Hospital Ethics Committee approved the study. A copy of the written consent is available for review by the Editor of this journal.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fan, J., Li, Y., Jia, R. et al. An inherited FGFR2 mutation increased osteogenesis gene expression and result in Crouzon syndrome. BMC Med Genet 19, 91 (2018). https://doi.org/10.1186/s12881-018-0607-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-018-0607-8