
Abstract
Background
In this study we investigated the association between SNPs in the S100B gene and Parkinson’s disease (PD) in two independent Swedish cohorts. The SNP rs9722 has previously been shown to be associated with higher S100B concentrations in serum and frontal cortex in humans. S100B is widely expressed in the central nervous system and has many functions such as regulating calcium homeostasis, inflammatory processes, cytoskeleton assembly/disassembly, protein phosphorylation and degradation, and cell proliferation and differentiation. Several of these functions have been suggested to be of importance for the pathophysiology of PD.
Methods
The SNPs rs9722, rs2239574, rs881827, rs9984765, and rs1051169 of the S100B gene were genotyped using the KASPar® PCR SNP genotyping system in a case-control study of two populations (431 PD patients and 465 controls, 195 PD patients and 378 controls, respectively). The association between the genotype and allelic distributions and PD risk was evaluated using Chi-Square and Cox proportional hazards test, as well as logistic regression. Linear regression and Cox proportional hazards tests were applied to assess the effect of the rs9722 genotypes on age of disease onset.
Results
The S100B SNPs tested were not associated with the risk of PD. However, in both cohorts, the T allele of rs9722 was significantly more common in early onset PD patients compared to late onset PD patients. The SNP rs9722 was significantly related to age of onset, and each T allele lowered disease onset with 4.9 years. In addition, allelic variants of rs881827, rs9984765, and rs1051169, were significantly more common in early-onset PD compared to late-onset PD in the pooled population.
Conclusions
rs9722, a functional SNP in the 3’-UTR of the S100B gene, was strongly associated with age of onset of PD.
Similar content being viewed by others

Background
Sporadic Parkinson’s disease (PD) or idiopathic PD is the far most common form of PD and accounts for at least 90% of all cases. Among the suggested pathophysiological mechanisms of neurodegeneration in PD are increased generation of reactive oxygen species, mitochondrial pathology, and increase in intracellular calcium [1,2,3]. There is also evidence that immune and inflammatory mechanisms as well as impaired protein degradation are involved in the pathogenesis [4,5,6]. Recently, it was suggested that sporadic PD could be due to misfolded alpha-synuclein that spreads and by change reaction induce misfolding and pathological aggregation of native alpha-synuclein [7,8,9]. Conceivably, these mechanisms may operate simultaneously or in time sequence.
Regarding sporadic PD, a number of genetic polymorphism-based studies has been performed on a variety of candidate genes (see http://www.pdgene.org) [10]. In a recently performed meta-analysis of genome-wide association studies (GWAS) significance was obtained for 28 gene loci [11]. However, according to genome-wide complex trait analysis there are substantially more risk loci to be identified [12].
S100B is a highly conserved protein and a member of the S100 calcium-binding protein superfamily. It is expressed in various cell types in the central nervous system, such as astrocytes, neural progenitor cells, and various neuronal populations [13, 14], as well as in the enteric nervous system in glial cells important for the regulation of inflammation in the gut [15]. Being both intracellularly and extracellularly active, S100B has a wide range of functions. Within cells, the protein regulates calcium homeostasis, cytoskeleton assembly/disassembly, protein phosphorylation and degradation, and cell proliferation and differentiation [16]. Secreted S100B have paracrine, autocrine, and endocrine properties, modulating the activity of neurons, astrocytes, microglia, monocytes, and endothelial cells [16].
Elevated serum concentrations of S100B have been detected in several pathological conditions, such as acute brain injuries [17], schizophrenia [18, 19], and Alzheimer’s disease [20]. Regarding PD, conflicting results have been obtained. In one study S100B serum concentrations were not significantly different between PD patients and controls [21]. In another study, however, antibodies against S100B were detected in the blood of PD patients, but not in the control group [22]. Sathe et al. [23] recently showed significantly higher S100B concentrations post-mortem in substantia nigra of PD patients.
Animal studies suggest S100B to be involved in motor and memory functions. Transgenic mice overexpressing S100B showed symptoms similar to PD, exhibiting impaired motor coordination [24], whereas S100B knock-out mice have exhibited enhanced spatial ability and synaptic plasticity [25].
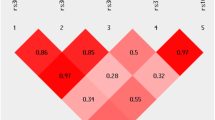
Considering these previous findings we performed a case-control study in two independent Swedish populations to evaluate the possible association between single nucleotide polymorphisms (SNPs) in the gene coding for S100B and PD. Since age of onset of PD seems to have a relatively high heritability, in one study estimated to be 40–60% [26], and previously have been reported to be associated with some gene polymorphisms [27], we also examined whether these SNPs affect age of onset of PD. We genotyped rs9722 and rs1051169 together with three other SNPs in the S100B gene, which were selected as Tag-SNPs (see Fig. 1).

LD plot generated for the S100B gene using HaploView v.4.2
Methods
Study populations
The discovery cohort consisted of 431 PD patients and 465 control subjects. The PD patients were recruited from four hospitals in Sweden (Gothenburg, Falköping, Skövde and Stockholm). Control subjects comprised unrelated outpatients in primary care in Gothenburg and participants in the Kungsholmen project, a community-based cohort in Stockholm of people aged 75 years and older [28]. Participants in the Kungsholmen project has been confirmed not to have PD. The validation cohort consisted of 195 PD patients and 378 control subjects. The PD patients were recruited from the hospitals in Linköping and Jönköping and control subjects were randomly collected from the population registry in the same recruitment area as for the hospitals. The patient and control groups in the validation cohort were frequency-matched by age and sex. All PD patients had been examined by neurologists and/or movement disorders specialists and fulfilled the Parkinson Disease Society Brain Bank criteria for idiopathic PD [29], except that the presence of more than one relative with the disease was not regarded as an exclusion criterion. Thirteen cases (3%) in the discovery cohort and 11 cases (5.6%) in the validation cohort had more than one affected relative. In the discovery cohort, 25% of the patients reported to have a first-, second- or third-degree relative with PD, and in the validation cohort, 21% of the patients report about having a first- or second degree relative with the disorder. Nearly all subjects (> 99%) were of Caucasian origin. All subjects had provided informed consent and the study were approved by the ethical committees at University of Gothenburg, Karolinska Institute and Linköping University.
Age of disease onset was defined as the time when the patients first noticed PD symptoms. The commonly used definition of having an “early age of onset” of PD if the disease begins at or before 50 years of age was used [6, 30, 31]. By using this definition, 87 patients (20%) in the discovery cohort and 25 patients (13%) in the validation cohort were categorized as having early-onset PD. The majority of the 87 early onset patients in the discovery cohort have previously been screened for mutations in the DJ-1, parkin, and PINK1 genes, and were not found be carriers of any of these [32, 33].
Genotyping and statistical analysis
DNA from blood samples were genotyped using the KASPar® PCR SNP genotyping system (KBiosciences, Herts, UK). The three tag-SNPs were chosen by pair wise tagging (r2 ≥ 0.80) from the International HapMap Project database (release 27, Phase II + III, February 2009, on NCBI B36 assembly, dbSNP b126). Thirty-eight individuals in the discovery cohort and four individuals in the validation cohort were excluded due to poor DNA quality. Success rates for the investigated SNPs were between 96.6–99.6%. Differences in allelic distributions were analysed using a Chi-square test in Haploview 4.0 (Broad Institute, Cambridge, MA, USA) and logistic regression in SPSS 19.0 (IBM Corporation, Armonk, NY, USA) and multiple testing correction was carried out on the pooled data by Bonferroni procedures (analyses of 5 SNPs and 3 comparisons: controls versus all PD patients, controls versus early onset PD and early onset versus late onset PD; 15 tests, corrected significance level: p = 0.0033). Cox proportional hazard tests were performed in SPSS 19.0 on the whole population including controls, as well as on patients only. Age at disease onset was used for patients and age at examination was used for controls. The association between the SNPs and age at disease onset was also evaluated using linear regression in SPSS. For both Cox proportional hazards analysis and linear regression analysis, gender and sample group (discovery cohort or validation cohort) were used as covariates. The significance level was set at p = 0.05.
Results
Demographic data of the populations are presented in Table 1. The genotype distributions of all five polymorphisms were in Hardy-Weinberg equilibrium in both control populations (p-value cut-off = 0.01). Three individuals were excluded in the statistical analysis of age of onset due to missing information about onset age. The allele and genotype distributions of the SNPs in Population 1 are displayed in Table 2.
In the discovery cohort, no significant differences in allelic or genotype frequencies were observed for any of the SNPs when comparing PD patients and controls, using Chi-square test and logistic regression analysis (results not shown). However, there were significant differences in allelic and genotype frequencies for several SNPs in the S100B gene when comparing PD patients with an early age of onset (≤50 years) to PD patients with a late disease onset as well as when comparing to controls. The T allele of rs9722, the C allele of rs9984765, and the G allele of rs1051169 were significantly more common in early-onset PD patients compared to late-onset PD patients (p = 0.0002, p = 0.008, and p = 0.037, respectively) and when compared to controls (p = 0.006, p = 0.005, and p = 0.035, respectively). Furthermore, the genotype frequencies of rs9722 and rs9984765 differed significantly when comparing early-onset PD patients and late-onset PD patients (p = 0.001 and p = 0.028, respectively) as well as when comparing early-onset PD patients and controls (p = 0.026 and p = 0.004, respectively).
To replicate these findings, we genotyped the S100B SNPs in an independent validation cohort (see Table 3). No significant differences in allele or genotype frequencies were observed for any of the SNPs when comparing all PD patients with controls in the validation cohort. However, the T allele of rs9722, and the C allele of rs881827 were significantly more common in early-onset than late-onset PD (p = 0.005 and p = 0.014, respectively) and the C allele of rs881827 was significantly more common in early-onset PD than in controls (p = 0.035). The genotype frequencies of rs9722 differed significantly when comparing early-onset to late onset PD patients (p = 0.021).
The allelic frequencies for the pooled populations are presented in Table 4, showing significant differences for all SNPs, except rs2239574. Notable is the highly significant allelic and genotype frequency difference of the rs9722 SNP in early-onset and late-onset PD (p = 0.0000041 and p = 0.00005, respectively (Bonferroni corrected p-values: p = 0.0000615 and p = 0.00075, respectively)). In line with these results, a Cox regression analysis comprising all PD patients in the pooled population (see Fig. 2) confirmed that the T-allele of rs9722 is associated with an earlier age of onset (HR = 1.49; 95% C.I = 1.17–1.90, p = 0.001). The Cox proportional hazards tests were insignificant when analyzing the whole population including controls (results not shown). Furthermore, when analyzing the data for the pooled population, linear regression showed that disease onset was significantly lower with more T alleles of the rs9722 polymorphism (p = 0.00004) and that each T allele lowered disease onset with 4.9 years. In addition, the haplotype TCCCG of the five genotyped SNPs (rs9722, rs2239574, rs881827, rs9984765, rs1051169) was also significantly more common in early- onset compared to late-onset PD in both populations (p = 0.01 for the discovery cohort, p = 0.035 for the validation cohort, and p = 0.000019 for both combined) as well as in early onset PD as compared to controls (p = 0.0099 for the discovery cohort, p = 0.064 for the validation cohort, and p = 0.0031 for both combined).

Age of onset of PD stratified by genotypes of the SNP rs9722, adjusted for gender and sample group
Discussion
In the present study rs9722, rs9984765 and rs10511669 in the S100B gene were associated with age of onset of PD in Population 1 and rs9722 and rs881827 were associated with age of onset in Population 2. In both populations, the T allele of rs9722 was strongly associated with early-onset PD. Furthermore, in both populations, the haplotype TCCCG of the five genotyped SNPs was more frequent in early-onset than in late-onset PD.
Several of the large GWAS studying PD [10] include SNPs in the S100B gene, although only one study genotyped the SNPs investigated in the present study [34]. However, other GWAS include SNPs in high LD with the ones genotyped in our study, which makes it possible to impute the genotypes of our SNPs. None of these studies report significant associations regarding PD and SNPs in the S100B gene, which is in line with the findings in the present study.
Noteworthy, only two GWAS so far have investigated age at onset of PD [35, 36]. In both studies, not rs9722 itself, but SNPs in high LD with rs9722, were genotyped and no significant association with age of onset were found. However, to be able to compare the results from different association studies it is important that the inclusion criteria for patients and controls used in the studies are similar, especially when searching for genetic risk variants of low impact in a complex disorder like PD. In the GWAS studying onset age in PD by Latourelle et al. [35], two of three of the PD populations investigated include samples recruited to study familial PD which means that all of the patients in those populations have a family history of the disease, making their sample different from ours where at least 80% of the patients are sporadic cases. Furthermore, in the GWAS by Spencer et al. [36], there is a quite large difference in mean age of onset (65.8 years) compared to our study (61.5 years). These dissimilarities might be part of the explanation to the deviation in results when comparing these two studies with the present one. Furthermore, the diversity of ethnicity might also be of importance and the populations studied in the present paper are very homogenous in that regard.
The S100B gene was investigated in a study of PD patients by Guo et al. [37]. The authors screened a Chinese PD-population for mutations in the coding parts of the gene, and consequently only one of the SNPs investigated in the present study, rs1051169, was possible to detect. The frequency found for this SNP was quite similar to the frequencies of it in our Caucasian PD patients.
It has been proposed that S100B has neurotrophic or neurotoxic properties depending on the extracellular concentration [16]. In normal conditions, S100B in nanomolar concentrations seems to protect neurons against oxidative stress [38, 39]. However, at higher extracellular concentrations, it may act as a pro-inflammatory substance activating astrocytes and microglia and inducing apoptosis [40,41,42]. Alternatively, S100B at high concentrations merely is a secondary reactive phenomena or marker of inflammation intensity rather than promoting inflammation (for discussion see Lam et al. [43]).
Parts of the effects of S100B appear to be mediated by the receptor for advanced glycation end products (RAGE) [44, 45]. In neurons, nanomolar concentrations of S100B promote cell survival by RAGE-mediated NF-KB activation, leading to upregulation of the anti-apoptotic factor Bcl-2 [39, 46, 47]. However, in micromolar concentrations, the RAGE-mediated S100B toxic effects are due to overproduction of reactive oxygen species (ROS) [44], leading to apoptosis.
The findings that high concentrations of S100B could have neurotoxic effects are especially interesting, because the rs9722 SNP, located in the 3′ untranslated region (3´-UTR), appears to be functional in that healthy individuals with the T allele variant, the variant we found to be more common in PD with early onset, have been reported to have higher serum and frontal cortex concentrations of S100B [48]. Furthermore, functional studies of peripheral blood mononuclear cells from healthy volunteers show that cells with the CT genotype of rs9722 express more than twice the amount of S100B mRNA as well as S100B protein as compared to cells with the CC genotype [49].
The allelic and genotype frequencies of the S100B polymorphisms were similar in late-onset PD and controls, but early-onset patients differed to both late-onset patients and controls. Interestingly, a newly published study analyzing data from a meta-analysis of GWAS found support for that an individual’s polygenic risk score were higher in PD patients with early onset as compared to those with late onset [50]. This pattern suggests that early-onset patients may have a different pathophysiology compared late-onset patients, with the S100B allelic variants conferring a risk only for early-onset patients. Support for this view comes from the observation that the incidence and prevalence of PD after the age of 50 increases almost exponentially in contrast to early onset PD [51], which besides is the basis for using this age as a cut-off while defining early age of onset PD.
However, another interpretation is that the functional activity of S100B does not influence the risk to be affected of neither PD with a late onset nor PD with an early onset but rather modulates the age of onset of PD, a notion that is further supported by the linear regression analysis and Cox proportional hazards tests performed in the present paper regarding the rs9722 SNP. Moreover, the observation that the genetic influence on the risk for sporadic PD, as judged by the very low concordance rate in monozygotic twins [52, 53], suggests that environmental factors are most important for causing the disease.
The distinction between gene variants influencing the risk to be affected by PD and variants that modify age of onset might be biologically significant; risk variants point to the initial cause, whereas onset modifiers implicate the processes that begin after the initial insult affecting the threshold for developing clinical signs [54]. Interestingly, segregation analyses of PD suggest stronger evidence for major genes influencing age of onset than for genes influencing susceptibility to disease [55, 56].
Conclusions
Even though the population sizes used in the current study are quite small, the results suggest that S100B activity could influence age of onset of sporadic PD. By resulting in higher S100B levels, the minor allele of the SNP rs9722 might modulate age of onset, potentially by activation of inflammatory processes or by increasing intracellular calcium.
Abbreviations
- GWAS:
-
Genome-wide Association Study
- PD:
-
Parkinson’s disease
- RAGE:
-
Glycation End Products
- SNP:
-
Single Nucleotide Polymorphism
References
Fahn S, Sulzer D. Neurodegeneration and neuroprotection in Parkinson disease. NeuroRx. 2004;1:139–54.
Corti O, Lesage S, Brice A. What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol Rev. 2011;91:1161–218.
Schapira AH. Mitochondrial pathology in Parkinson’s disease. Mount Sinai J Med. 2011;78:872–81.
Barnum CJ, Tansey MG. Modeling neuroinflammatory pathogenesis of Parkinson's disease. Prog Brain Res. 2010;184:113–32.
Håkansson A, Westberg L, Nilsson S, Buervenich S, Carmine A, Holmberg B, et al. Investigation of genes coding for inflammatory components in Parkinson's disease. Mov Disord. 2005;20:569–73.
Håkansson A, Westberg L, Nilsson S, Buervenich S, Carmine A, Holmberg B, et al. Interaction of polymorphisms in the genes encoding interleukin-6 and estrogen receptor beta on the susceptibility to Parkinson's disease. Am J Med Genet B Neuropsychiatr Genet. 2005;133B:88–92.
Lerner A, Bagic A. Olfactory pathogenesis of idiopathic Parkinson disease revisited. Mov Disord. 2008;23:1076–84.
Dunning CJ, Reyes JF, Steiner JA, Brundin P. Can Parkinson’s disease pathology be propagated from one neuron to another? Prog Neurobiol. 2012;97:205–19.
Hilker R, Brotchie JM, Chapman J. Pros and cons of a prion-like pathogenesis in Parkinson's disease. BMC Neurol. 2011;11:74.
PDGene database. http://www.pdgene.org
Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat Genet. 2014;46:989–93.
Keller MF, Saad M, Bras J, Bettella F, Nicolaou N, Simón-Sánchez J, et al. Using genome-wide complex trait analysis to quantify ‘missing heritability’ in Parkinson's disease. Hum Mol Genet. 2012;21:4996–5009.
Rickmann M, Wolff JR. S100 protein expression in subpopulations of neurons of rat brain. Neurosci. 1995;67:977–91.
Vives V, Alonso G, Solal AC, Joubert D, Legraverend C. Visualization of S100B-positive neurons and glia in the central nervous system of EGFP transgenic mice. J Comp Neurol. 2003;457:404–19.
Cirillo C, Sarnelli G, Esposito G, Turco F, Steardo L, Cuomo R. S100B protein in the gut: the evidence for enteroglial-sustained intestinal inflammation. World J Gastroenterol. 2011;17:1261–6.
Donato R, Sorci G, Riuzzi F, Arcuri C, Bianchi R, Brozzi F, et al. S100B’s double life: intracellular regulator and extracellular signal. Biochim Biophys Acta. 2009;1793:1008–22.
Rothoerl RD, Woertgen C, Holzschuh M, Metz C, Brawanski A. S-100 serum levels after minor and major head injury. J Trauma. 1998;45:765–7.
Rothermundt M, Ponath G, Glaser T, Hetzel G, Arolt V. S100B serum levels and long-term improvement of negative symptoms in patients with schizophrenia. Neuropsychopharmacology. 2004;29:1004–11.
Schmitt A, Bertsch T, Henning U, Tost H, Klimke A, Henn FA, et al. Increased serum S100B in elderly, chronic schizophrenic patients: negative correlation with deficit symptoms. Schizophr Res. 2005;80:305–13.
Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, et al. Brain interleukin 1 and S-100 immunoreactivity are elevated in down syndrome and Alzheimer disease. Proc Natl Acad Sci U S A. 1989;86:7611–5.
Schaf DV, Tort AB, Fricke D, Schestatsky P, Portela LV, Souza DO, et al. S100B and NSE serum levels in patients with Parkinson’s disease. Parkinsonism Relat Disord. 2005;11:39–43.
Wilhelm KR, Yanamandra K, Gruden MA, Zamotin V, Malisauskas M, Casaite V, et al. Immune reactivity towards insulin, its amyloid and protein S100B in blood sera of Parkinson's disease patients. Eur J Neurol. 2007;14:327–34.
Sathe K, Maetzler W, Lang JD, Mounsey RB, Fleckenstein C, Martin HL, et al. S100B is increased in Parkinson's disease and ablation protects against MPTP-induced toxicity through the RAGE and TNF-alpha pathway. Brain. 2012;135:3336–47.
Liu J, Wang H, Zhang L, Xu Y, Deng W, Zhu H, et al. S100B transgenic mice develop features of Parkinson's disease. Arch Med Res. 2011;42:1–7.
Nishiyama H, Knopfel T, Endo S, Itohara S. Glial protein S100B modulates long-term neuronal synaptic plasticity. Proc Natl Acad Sci U S A. 2002;99:4037–42.
Li Y-J, Scott WK, Hedges DJ, Zhang F, Gaskell PC, Nance MA, et al. Age at onset in two common neurodegenerative diseases is genetically controlled. Am J Hum Gen. 2002;70:985–93.
Searles Nielsen S, Bammler TK, Gallagher LG, Farin FM, Longstreth W Jr, Franklin GM, et al. Genotype and age at Parkinson disease diagnosis. Int J Mol Epidemiol Genet. 2013;4:61–9.
Fratiglioni L, Viitanen M, Backman L, Sandman PO, Winblad B. Occurrence of dementia in advanced age: the study design of the Kungsholmen project. Neuroepidemiology. 1992;11(Suppl 1):29–36.
Daniel SE, Lees AJ. Parkinson's disease society brain Bank, London: overview and research. J Neural Transm Suppl. 1993;39:165–72.
Mizuta I, Nishimura M, Mizuta E, Yamasaki S, Ohta M, Kuno S, et al. Relation between the high production related allele of the interferon-gamma (IFN-gamma) gene and age at onset of idiopathic Parkinson's disease in Japan. J Neurol Neurosurg Psychiatry. 2001;71:818–9.
Wang J, Zhao CY, Si YM, Liu ZL, Chen B, Yu L. ACT and UCH-L1 polymorphisms in Parkinson's disease and age of onset. Mov Disord. 2002;17:767–71.
Håkansson A, Belin AC, Stiller C, Sydow O, Johnels B, Olson L, et al. Investigation of genes related to familial forms of Parkinson’s disease – with focus on the Parkin gene. Parkinsonism Relat Disord. 2008;14:520–2.
Anvret A, Blackinton JG, Westerlund M, Ran C, Sydow O, Willows T, et al. DJ-1 mutations are rare in a Swedish Parkinson cohort. Open Neurol J. 2011;5:8–11.
Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson's disease. Nat Genet. 2010;42:781–5.
Latourelle JC, Pankratz N, Dumitriu A, Wilk JB, Goldwurm S, Pezzoli G, et al. Genomewide association study for onset age in Parkinson disease. BMC Med Genet. 2009;10:98. BMC Medical Genetics 2009. https://doi.org/10.1186/1471-2350-10-98.
Consortium, U. K. P. S. D., Wellcome Trust Case Control, C, Spencer CC, Plagnol V, Strange A, Gardner M, Paisan-Ruiz C, Band G, et al. Dissection of the genetics of Parkinson's disease identifies an additional association 5′ of SNCA and multiple associated haplotypes at 17q21. Hum Mol Genet. 2011;20:345–53.
Guo Y, Yang H, Deng X, Song Z, Yang Z, Xiong W, et al. Genetic analysis of the S100B gene in Chinese patients with Parkinson disease. Neurosci Lett. 2013;555:134–6.
Winningham-Major F, Staecker JL, Barger SW, Coats S, Van Eldik LJ. Neurite extension and neuronal survival activities of recombinant S100 beta proteins that differ in the content and position of cysteine residues. J Cell Biol. 1989;109:3063–71.
Haglid KG, Yang Q, Hamberger A, Bergman S, Widerberg A, Danielsen N. S-100beta stimulates neurite outgrowth in the rat sciatic nerve grafted with acellular muscle transplants. Brain Res. 1997;753:196–201.
Li Y, Barger SW, Liu L, Mrak RE, Griffin WS. S100beta induction of the proinflammatory cytokine interleukin-6 in neurons. J Neurochem. 2000;74:143–50.
Adami C, Sorci G, Blasi E, Agneletti AL, Bistoni F, Donato R. S100B expression in and effects on microglia. Glia. 2001;33:131–42.
Bianchi R, Kastrisianaki E, Giambanco I, Donato R. S100B protein stimulates migration via RAGE-dependent up-regulation of chemokine expression and release. J Biol Chem. 2011;286:7214–26.
Lam V, Albrecht MA, Takechi R, Giles C, James AP, Foster JK, et al. The serum concentration of the calcium binding protein S100B is positively associated with cognitive performance in older adults. Front Aging Neurosci. 2013;5:61.
Huttunen HJ, Kuja-Panula J, Sorci G, Agneletti AL, Donato R, Rauvala H. Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J Biol Chem. 2000;275:40096–105.
Villarreal A, Aviles Reyes RX, Angelo MF, Reines AG, Ramos AJ. S100B alters neuronal survival and dendrite extension via RAGE-mediated NF-kappaB signaling. J Neurochem. 2011;117:321–32.
Ahlemeyer B, Beier H, Semkova I, Schaper C, Krieglstein J. S-100beta protects cultured neurons against glutamate- and staurosporininduced damage and is involved in the anti-apoptotic action of the 5 HT(1A)-receptor agonist, bay x 3702. Brain Res. 2000;858:121–8.
Businaro R, Leone S, Fabrizi C, Sorci G, Donato R, Lauro GM, et al. S100B protects LAN-5 neuroblastoma cells against Abeta amyloid-induced nheurotoxicity via RAGE engagement at low doses but increases Abeta amyloid neurotoxicity at high doses. J Neurosci Res. 2006;83:897–906.
Hohoff C, Ponath G, Freitag CM, Kästner F, Krakowitzky P, Domschke K, et al. Risk variants in the S100B gene predict elevated S100B serum concentrations in healthy individuals. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:291–7.
Cunha C, Giovannini G, Pierini A, Bell AS, Sorci G, Riuzzi F, et al. Genetically-determined hyperfunction of the S100B/RAGE axis is a risk factor for aspergillosis in stem cell transplant recipients. PLoS One. 2011;6:e27962.
Escott-Price V, Nalls MA, Morris HR, Lubbe S, Brice A, Gasser T, et al. Polygenic risk of Parkinson disease is correlated with disease age at onset. Ann Neurol. 2015;77:582–91.
Tanner CM, Goldman SM. Epidemiology of Parkinson's disease. Neurol Clin. 1996;14:317–35.
Wirdefeldt K, Gatz M, Schalling M, Pedersen NL. No evidence for heritability of Parkinson disease in Swedish twins. Neurol. 2004;6:305–11.
Wirdefeldt K, Gatz M, Reynolds CA, Prescott CA, Pedersen NL. Heritability of Parkinson disease in Swedish twins: a longitudinal study. Neurobiol Aging. 2011;32:1923e1–8.
Payami H, Kay DM, Zabetian CP, Schellenberg GD, Factor SA, McCulloch CC. Visualizing disease associations: graphic analysis of frequency distributions as a function of age using moving average plots (MAP) with application to Alzheimer's and Parkinson's disease. Genet Epidemiol. 2010;34:92–9.
Zareparsi S, Taylor TD, Harris EL, Payami H. Segregation analysis of Parkinson disease. Am J Med Genet. 1998;80:410–7.
Maher NE, Currie LJ, Lazzarini AM, Wilk JB, Taylor CA, Saint-Hilaire MH, et al. Segregation analysis of Parkinson disease revealing evidence for a major causative gene. Am J Med Genet. 2002;109:191–7.
Acknowledgements
We would like to thank Professor Laura Fratiglioni for providing us with control samples from the Swedish National Study on Aging and Care in Kungsholmen (SNAC-K).
Funding
The study was supported by the Swedish Research Council, Swedish Brain Power, The LUA-ALF Foundation at the Sahlgrenska University Hospital, Swedish Parkinson Foundation, Karolinska Institutet Funds, Åke Wibergs Stiftelse, and Foundation for Parkinson Research in Linköping.
Availability of data and materials
The datasets used during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
CF and AZ substantially contributed to the design of the study, acquisition of data, data analysis and manuscript writing and revising. CR, ACB, OS, LB, BH, ND and PS carried out the recruitment of participants and collection of samples. AE contributed substantially to the conception and design of the study. HN directed the project and substantially contributed to the design of the study, acquisition of data, data analysis and manuscript writing and revising. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All subjects provided written informed consent and the study were approved by the ethical committees at University of Gothenburg, Karolinska Institute and Linköping University.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fardell, C., Zettergren, A., Ran, C. et al. S100B polymorphisms are associated with age of onset of Parkinson’s disease. BMC Med Genet 19, 42 (2018). https://doi.org/10.1186/s12881-018-0547-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-018-0547-3