
Abstract
Background
MYO15A variants, except those in the N-terminal domain, have been shown to be associated with congenital or pre-lingual severe-to-profound hearing loss (DFNB3), which ultimately requires cochlear implantation in early childhood. Recently, such variants have also been shown to possibly cause moderate-to-severe hearing loss. Herein, we also demonstrate that some MYO15A mutant alleles can cause postlingual onset of progressive partial deafness.
Methods
Two multiplex Korean families (SB246 and SB224), manifesting postlingual, progressive, partial deafness in an autosomal recessive fashion, were recruited. Molecular genetics testing was performed in two different pipelines, in a parallel fashion, for the SB246 family: targeted exome sequencing (TES) of 129 known deafness genes from the proband and whole exome sequencing (WES) of all affected subjects. Only the former pipeline was performed for the SB224 family. Rigorous bioinformatics analyses encompassing structural variations were executed to investigate any causative variants.
Results
In the SB246 family, two different molecular diagnostic pipelines provided exactly the same candidate variants: c.5504G > A (p.R1835H) in the motor domain and c.10245_10247delCTC (p.S3417del) in the FERM domain of MYO15A. In the SB224 family, c.9790C > T (p.Q3264X) and c.10263C > G (p.I3421M) in the FERM domain were detected as candidate variants.
Conclusions
Some recessive MYO15A variants can cause postlingual onset of progressive partial deafness. The phenotypic spectrum of DFNB3 should be extended to include such partial deafness. The mechanism for a milder phenotype could be due to the milder pathogenic potential from hypomorphic alleles of MYO15A or the presence of modifier genes. This merits further investigation.
Similar content being viewed by others

Background
MYO15A, a causative gene of DFNB3 (OMIM 600316) [1], is a frequently detected deafness gene. Friedman et al., in Bali, first reported that the frequency of autosomal recessive hearing loss caused by the MYO15A pathogenic variant was about 2% [2]. Thereafter, the frequency of MYO15A pathogenic variant was reported in up to 9.9% of deafness cases in Turkey [3]. In our previous study, MYO15A pathogenic variant was reported at a frequency of 2.1% in nonsyndromic autosomal recessive deafness, which was the fourth most common deafness gene in Korea, following SLC26A4, GJB2 and CDH23 [4]. Sequentially, several studies have been performed to investigate the effects of MYO15A pathogenic variants on hearing loss [5,6,7,8,9].
The role of myosin XVA, which is encoded by MYO15A, includes the graded elongation and maintenance of stereocilia and actin-organization in the inner ear hair cells. These are both essential for normal auditory function. Therefore, MYO15A pathogenic variants were initially thought to induce congenital severe-to-profound hearing loss [1, 10,11,12,13,14,15]. However, it was later discovered that the phenotypes of MYO15A pathogenic variants varied depending on the affected domain. The variation of phenotypes according to the affected domain has been explained by the existence of two isoforms -- a class 1 isoform with the N-terminal domain, which is encoded by exon 2, and a class 2 isoform with no N-terminal domain [16, 17]. The pathogenic variant in N-terminal domain affects only the class 1 isoform without affecting the class 2 isoform [18]. The class 2 isoform is present in the human inner ear [17]. Therefore, pathogenic variants in the N-terminal domain are known to cause minor deficiencies in the inner ear, resulting in amilder auditory phenotype, when compared with other pathogenic variants in MYO15A [5,6,7]. Conversely, MYO15A pathogenic variants that reside in the regions shared by both isoforms are known to cause congenital or prelingual severe-to-profound hearing loss [7].
Recently, Naz et al. reported that MYO15A pathogenic variants, which had previously been thought to only cause profound hearing loss, may cause moderate-to-severe hearing loss [9]. We also found two families with MYO15A pathogenic variants in the motor and FERM domains, which were expected to cause profound hearing loss. They showed postlingual onset of bilateral symmetrical, partial deafness with significant residual hearing at low frequencies. Based on these results, we suggest that MYO15A may be a causative gene responsible for the postlingual onset of progressive partial deafness, which in turn requires the expansion of the phenotypic spectrum of MYO15A pathogenic variants.
Methods
Human subjects
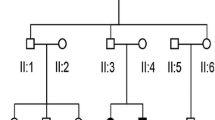
All procedures in this study were approved by the Institutional Review Boards of Seoul National University Hospital (IRBY-H-0905-041-281) and Institutional Review Boards of Seoul National University Bundang Hospital (IRB-B-1007-105-402). Written informed consent was obtained from all participants. Two multiplex Korean families (SB246 and SB224), with the segregation of postlingual, bilaterally symmetrical, partial hearing loss in an autosomal recessive fashion, were included in this study. The first family (SB246) was comprised of five individuals, three of whom participated in the study; the second family (SB224) was comprised of four individuals, all of whom participated. Two generations were included in each family (Fig. 1). Phenotypic evaluations included medical and developmental history interviews, physical examinations, and audiometric evaluation.

Pedigree and pure-tone audiograms of family SB246 (a) and 224 (b). SB246–482 and SB224–437 showed bilateral, severe, and symmetrical SNHL. SB246–483 and 484 showed bilateral, moderate, and symmetrical SNHL. In all subjects, hearing at low frequencies was significantly preserved. SB224–437 had cochlear implantation at the age of 20. During the operation, insertion of Med-El device, Flex 24 (Angular insertion depth [28] of 393°) into the cochlea was performed, and residual hearing of low frequencies was substantially preserved. *, subjects who underwent targeted exome sequencing of 129 known deafness genes and whole exome sequencing; †, subjects who underwent targeted exome sequencing of 129 known deafness
Audiometric evaluation
Pure-tone audiometry was performed on SB246–482, 483, and 484 as well as on SB224–437, in accordance with the standard protocols. The air and bone conduction thresholds were obtained at frequencies of 250–8000 Hz. The hearing loss range was divided into three parts: low frequency, 250–500 Hz; mid frequency, 1–2 kHz; and high frequency, 4–8 kHz [19]. The mean levels of hearing loss for all frequencies as well as low, mid, and high frequencies were calculated.
Molecular genetic test
In the SB246 family, molecular genetic testing was performed in two different pipelines, in a parallel fashion. The first pipeline was targeted exome sequencing (TES) of 129 known deafness genes from the proband, followed by a segregation study using Sanger sequencing; the second pipeline was whole exome sequencing (WES) in the three affected subjects. In the SB224 family, TES of 129 known deafness genes was performed from the proband, followed by a segregation study using Sanger sequencing.
Targeted exome sequencing
TES and bioinformatics analyses were performed, as previously described [20,21,22,23,24,25]. The DNA samples from SB246–482 and SB224–437 underwent TES of 129 known deafness genes (TRS-129) by Otogenetics (Norcross, GA, USA) (Additional file 1: Table S1). The acquired reads were mapped onto the UCSC hg19 reference genome assembly, using Lasergene 14 software package (DNASTAR, Madison, WI, USA) (Additional file 2: Table S2). Further bioinformatics analyses were performed to identify all variants. As a basic filtering step, non-synonymous single nucleotide polymorphisms (SNPs) with read depths ≥20 were chosen. The known disease-causing SNPs or SNPs with global minor allele frequency (MAF) ≤ 0.002 were chosen. Global MAF was checked using several databases, including 1000 Genomes, Exome Aggregation Consortium (ExAC), and NHLBI Grand Opportunity Exome Sequencing Project (GO-ESP). These non-synonymous SNPs were compared against the Korean Reference Genome Database (KRGDB), that consists of 622 Korean individuals (1,244alleles) (http://152.99.75.168/KRGDB/menuPages/firstInfo.jsp). SNPs with allele frequency < 0.005 were chosen. Inheritance patterns were checked and SNPs that did not coincide with the autosomal recessive pattern were excluded. To predict the pathogenicity of each variant, SIFT, PolyPhen-2 analyses, and Pathogenic variantTaster were performed. Pathogenic variants are described in the context of the American College of Medical Genetics and Genomics (ACMG) 2015 guidelines [26]. The evolutionary conservation of the amino acid sequence was estimated using the GERP++ score in the UCSC Genome Browser (http://genome.ucsc.edu/). The variants that were predicted as benign by silico prediction were excluded. The remaining SNPs were validated in other family members (SB246–483, 484, 485 and 486 and SB224–592, 593 and 594) by Sanger sequencing.
Whole exome sequencing
DNA samples from SB246–482, 483, and 484 were subjected to WES by Macrogen (Seoul, South Korea) (Additional file 3: Table S3). First, we filtered out the variants in non-coding regions, as well as synonymous variants in coding regions. Second, the variants with MAF < 1% were selected based on Exome Sequencing Project 6500 (ESP6500), 1000 Genomes Project (1000G), ExAC, and our in-house database containing the exomes of 192 Korean individuals. Third, following the inheritance pattern, homozygous variants and compound heterozygote variants with enough read depths (> 10×) and a genotype quality (> 20) were selected. Finally, to exclude the variants without clinical significance, flagged SNPs based on dbSNP ID (dbSNP 147) were selected. The relation of selected genes with diseases or functions was identified through previous studies. The variants related to hearing loss were selected. Moreover, rigorous bioinformatics analyses encompassing the structural variations were performed to investigate the causative variants. Copy number variations (CNV) were calculated by EXCAVATOR2 [27] with 20 k window size; the pooling mode and in-house CNV tool were used to estimate the log2 normalized depth ratio of each targeted region. Finally, we selected CNVs containing hearing loss gene or locus.
Results
Auditory phenotype
Pure-tone audiograms for the affected individuals are presented at Fig. 1. SB246–482 and SB224–437 showed bilateral, severe, and symmetrical sensorineural hearing loss (SNHL). SB224–437 showed a positive stapedial reflex at the time of toddler, suggesting that the subject did not have severe SNHL at that time (data not shown). Due to the progression of SNHL into a severe degree, especially in mid-to-high frequencies, SB224–437 underwent cochlear implantation in his/her twenties. During the operation, an insertion of Med-El device, Flex 24 (Angular insertion depth [28] of 393°) rather than Flex 28 into the cochlea, was performed, aiming to preserve low frequency hearing. Consequently, residual hearing of low frequencies was preserved substantially (Fig. 1b). Pure-tone audiometry was not performed for SB224–592 at our hospital. However, the pure-tone audiogram from another hospital showed that SB224–592’s hearing thresholds were 75 dB HL on both sides, and SB224–592 used bilateral hearing aids.
Interestingly, SB246–483 and 484 showed bilateral partial deafness, characterized by normal thresholds in low frequencies, but with severe SNHL in high frequencies. In SB246–482 and SB224–437, the mean threshold level for high frequencies was the highest, followed by mid and low frequencies. In all subjects, hearing at low frequencies was significantly preserved.
Targeted exome sequencing
TES was performed in SB246–482 and SB224–437. The reads were aligned to the human reference genome (GRCh37/hg19), using SeqMan NGen, and the targeted variants were analyzed using ArrayStar software (version 14.1.0; DNASTAR, WI, USA). Bioinformatics analyses were performed (Fig. 2a). After the basic filtering step, eight and six variants were selected as candidate pathogenic variants for SB246–482 and SB224–437, respectively.

Schematic flowchart of filtering of causative variants in this study. a Targeted exome sequencing. b Whole exome sequencing. AR, autosomal recessive; AD, autosomal dominant
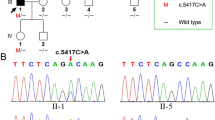
As the inheritance pattern was autosomal recessive, variants that did not follow an autosomal recessive inheritance pattern were excluded. Through in silico prediction, a benign variant was excluded. Lastly, candidate variants were validated using Sanger sequencing in parents (SB246–485 and 486) and siblings (SB246–483 and 484) of SB246–482, as well as in parents (SB224–593 and 594) and sibling (SB224–592) of SB224–437. Only two candidate variants from a single gene, MYO15A, survived the filtering steps in each family (Fig. 2a). In the SB246 family, c.5504G > A (p.R1835H) and a single aminoacid deletion, c.10245_10247delCTC (p.S3417del), remained after the final filtering step (Table 1 and Fig. 3). The parents (SB246–485 and 486) of SB246–482 were heterozygous for c.5504G > A (p.R1835H) and c.10245_10247delCTC (p.S3417del), respectively (Fig. 4a). Moreover, p.R1835 and p.S3417 were well-conserved in several species, and in silico prediction supported the pathogenicity of these variants. In the SB224 family, a nonsense variant, c.9790C > T (p.Q3264X), and a missense variant, c.10263C > G (p.I3421M), remained (Table 1). The parents (SB224–593 and 594) of SB224–437 were heterozygous for c.9790C > T (p.Q3264X) and c.10263C > G (p.I3421M), respectively (Fig. 4b). Furthermore, p.Q3264 and p.I3421 were well-conserved in several species, and in silico prediction supported the pathogenicity of these variants.

The locations of MYO15A variants

Segregation of MYO15A variants in two families, SB246 and 224. a-c Sanger sequencing traces for c.5504G > A (p.R1835H) + c.10245_10247delCTC (p.S3417del) compound heterozygote (SB246–482, 483 and 484). d Sanger sequencing traces for c.5504G > A carrier (SB246–485). e Sanger sequencing traces for c.10245_10247delCTC carrier (SB246–486). f Conservation of mutant residues among the orthologs from several species; p.R1835 and p.S3417 are conserved among all species, ranging from humans to zebrafish. g and h Sanger sequencing traces for c.9790C > T (p.Q3264X) + c.10263C > G (p.I3421M) compound heterozygote (SB224–437 and 592). i Sanger sequencing traces for c.9790C > T carrier (SB224–593). j Sanger sequencing traces for c.10263C > G carrier (SB224–594). k Conservation of mutant residues among the orthologs from several species; p.Q3264 and p.I3421 are conserved among all species, ranging from humans to zebrafish
Whole exome sequencing
Independently of molecular genetic testing using TES, WES of genomic DNA from three affected subjects (SB246–482, 483 and 484) in the SB246 family was performed in a parallel fashion. Following the basic filtering step, 1188 variants from 895 genes were selected as candidate pathogenic variants (Fig. 2b). The inheritance pattern was regarded as autosomal recessive, and variants that did not fit for this inheritance pattern were excluded. After excluding the variants without clinical significance, seven variants from five genes remained (Fig. 2b and Table 2). Among the five genes, only MYO15A was related to hearing loss (Table 2). Two candidate variants, c.5504G > A and c.10245_10247delCTC from a single gene, MYO15A, remained. In addition, a large heterozygous genomic deletion involving STRC was detected from SB246–482; however, it did not segregate among the other two affected subjects. In the evaluation of CNV, one copy number alteration residing in STRC and CATSPER2, which are both known hearing loss genes, was detected by both EXCAVATOR2 and in-house CNV tool from only SB246–482 (Fig. 5a, b). The deletion of STRC and CATSPER2 of SB246-482, if in a homozygous form, could be pathogenic, leading to dieafness-infertility syndrome.

Copy number variant identification by in-house CNV tool and EXCAVATOR2 with WES. a The horizontal axis indicates chromosomal position, and the vertical axis indicates the log2 normalized depth ratio; 0 indicates 2 copies and − 1.0 indicates 1 copy deletion, respectively. The red line indicates SB246–482, light-blue line indicates SB246–483, and green line indicates SB246–484, respectively. A heterozygote deletion in the STRC and CATSPER2 region is seen in only SB246–482. The hypothesized mechanisms for the event is shown to the right. b The result of EXCAVATOR2 also indicates 1 copy deletion in the region where STRC and CATSPER2 as well as PPIP5K1 and CKMT1A were located in
Discussion
Although TES of 129 genes in the deafness panel is an efficient and convenient tool for detecting known causative variants, pathogenic variants from a novel deafness gene may not be detected using only TES. To explore and minimize such a possibility, we performed genetic testing via two diagnostic pipelines, TES and WES, in a parallel fashion for the SB246 family. Filtering of candidate variants through these two pipelines indicated the same result, strongly supporting our molecular diagnosis. Moreover, certain genes, such as OTOF and STRC, have been reported not to be fully covered by next generation sequencing [29, 30]. To overcome this, we modified the mapping quality of bioinformatics analysis. Furthermore, we excluded the possibility of structural variations involving STRC and CATSPER2, which was reported to be an important molecular etiology of SNHL in Japan [30]. In the SB224 family, two variants, c.9790C > T (p.Q3264X) and c.10263C > G (p.I3421M), of MYO15A survived at the final filtering step of TES analyses; c.9790C > T was a nonsense variant causing truncation of the protein, and c.10263C > G has already been reported to be pathogenic. Therefore, it is most likely that hearing loss from SB224–437 was attributed to these two MYO15A variants. Sequentially, WES was not performed in SB224.
The conservation and pathogenicity prediction study also strengthened our hypothesis that MYO15A pathogenic variants, c.5504G > A (p.R1835H) and c.10245_10247delCTC (p.S3417del), in the SB246 family and c.9790C > T (p.Q3264X) and c.10263C > G (p.I3421M), in the SB224 family were the causative pathogenic variants of hearing loss. Among the four MYO15A pathogenic variants discovered here, c.5504G > A and c.9790C > T were novel variants; c.9790C > T was a nonsense pathogenic variant, and c.5504G > A resided in the motor domain. The motor domain is one of the most important domains in myosin XVA. Sequentially, it is reasonable to infer that variants in the motor domain may lead to profound hearing loss. c.10245_10247delCTC and c.10263C > G resided in the FERM domain; they have already been reported to cause congenital profound hearing loss [31,32,33]. Therefore, four variants detected in this study were expected to cause profound hearing loss; however, they were associated with partial deafness with significant residual or even near-normal hearing at low frequencies.
The establishment of genotype-phenotype correlation is one of the fundament goals of genetics, as it enables personalized and timely management of diseases, leading to great contribution to precision medicine. Personalized and timely auditory rehabilitation is crucial in hearing loss because there is a critical time window for auditory development and degeneration. Therefore, an establishment of genotype-phenotype correlation is a meaningful issue in hearing loss. Sequentially, genotype-phenotype correlations of several genes and their pathogenic variants have been investigated widely. However, genotype-phenotype correlations have generally been based on the observation of phenotypes, rather than prudent analysis of pathogenic mechanism. Previously established genotype-phenotype correlations could be modified. Recently, several genes, including MYO15A, CDH23, and PTPRQ, have been reported to cause different types of hearing loss from those previously postulated in the literature [9, 31].
MYO15A is one of the genes with a well-documented genotype-phenotype correlation. It has been reported that hearing loss phenotype related to MYO15A is different according to the affected domain. Specifically, pathogenic variants in the N-terminal domain of MYO15A have been suggested to be associated with residual hearing [5,6,7,8], especially at low frequencies, while pathogenic variants in other domains resulted in congenital severe-to-profound hearing loss [1, 10,11,12,13,14,15]. With the progress of various auditory rehabilitation technologies, appropriate auditory rehabilitation tailored to the type of hearing loss should be implemented. A moderate hearing loss can be fully rehabilitated with hearing aids; however, cochlear implantation may be necessary in cases of severe hearing loss. Moreover, for subjects with overall profound hearing loss, while retaining significant residual hearing at low frequencies, electroacoustic stimulation (EAS) may be the best option. Therefore, a prediction of phenotypes in accordance with the affected domain in MYO15A has greatly contributed to personalized and timely auditory rehabilitation. Recently, these previously established genotype-phenotype correlations appear to require an updated modification. Some MYO15A pathogenic variants affecting the domains other than the N-terminal, have shown to cause moderate-to-severe hearing loss, not profound hearing loss [9]. We also found two families carrying MYO15A pathogenic variants, which were expected to cause congenital severe-to-profound hearing loss, but resulted in postlingual onset of progressive partial deafness with residual hearing at low frequencies. Indeed, SB224–437 carrying two MYO15A mutant alleles in this current study had a shallower angular insertion depth of 393°, by Flex 24 rather than by the longer Flex 28 electrode to minimize the shift in low frequency thresholds from this subject (Fig. 1b).
A milder phenotype may have been influenced by factors like milder pathogenic potential from hypomorphic alleles of MYO15A, genetic modifiers that reduce severity of hearing loss, or environmental factors. A recent progress in the genetic diagnosis technique can also contribute to the expansion of phenotypic spectrum of MYO15 alterations. In the past, genetic hearing loss had been found in consanguineous families using a linkage analysis, especially homozygous mapping. Therefore, it is likely that the severe phenotypes caused by severely pathogenic homozygous pathogenic variants had been preferentially recruited for the genetic study. However, with the development of next generation sequencing, there has been an increase in the frequency of molecular diagnostic testing of small-to-mid sized, non-consanguineous families, leading to the emergence of many compound heterozygotes with varying degrees of pathogenicity. Consequently, it has become possible to discover various phenotypes from the existing genes. Further studies are needed about other potential factors affecting phenotypes, such as genetic modifiers or environmental factors.
Conclusions
We discovered that four MYO15A pathogenic variants in the motor and FERM domains caused partial deafness with significant residual hearing at low frequencies. This is interesting since these pathogenic variants were previously thought to cause profound hearing loss, without any association with partial deafness. This result suggests that some MYO15A variants may cause adult-onset, progressive partial deafness. The phenotypic spectrum of DFNB3 should be extended to include such partial deafness.
Abbreviations
- 1000G:
-
1000 Genomes Project
- CNV:
-
Copy number variation
- EAS:
-
Electroacoustic stimulation
- ESP6500:
-
Exome Sequencing Project 6500
- ExAC:
-
Exome Aggregation Consortium
- GO-ESP:
-
Grand Opportunity Exome Sequencing Project
- KRGDB:
-
Korean Reference Genome Database
- MAF:
-
Minor allele frequency
- SNHL:
-
Sensorineural hearing loss
- SNP:
-
Single nucleotide polymorphism
- TES:
-
Targeted exome sequencing
- WES:
-
Whole exome sequencing
References
Wang A, Liang Y, Fridell RA, Probst FJ, Wilcox ER, Touchman JW, Morton CC, Morell RJ, Noben-Trauth K, Camper SA, et al. Association of unconventional myosin MYO15 mutations with human nonsyndromic deafness DFNB3. Sci NY. 1998;280(5368):1447–51.
Friedman TB, Liang Y, Weber JL, Hinnant JT, Barber TD, Winata S, Arhya IN, Asher JH Jr. A gene for congenital, recessive deafness DFNB3 maps to the pericentromeric region of chromosome 17. Nat Genet. 1995;9(1):86–91.
Duman D, Sirmaci A, Cengiz FB, Ozdag H, Tekin M. Screening of 38 genes identifies mutations in 62% of families with nonsyndromic deafness in Turkey. Genet Test Mol Biomarkers. 2011;15(1–2):29–33.
Park JH, Kim NK, Kim AR, Rhee J, Oh SH, Koo JW, Nam JY, Park WY, Choi BY. Exploration of molecular genetic etiology for Korean cochlear implantees with severe to profound hearing loss and its implication. Orphanet J Rare Dis. 2014;9:167.
Bashir R, Fatima A, Naz S. Prioritized sequencing of the second exon of MYO15A reveals a new mutation segregating in a Pakistani family with moderate to severe hearing loss. Eur J Med Genet. 2012;55(2):99–102.
Cengiz FB, Duman D, Sirmaci A, Tokgoz-Yilmaz S, Erbek S, Ozturkmen-Akay H, Incesulu A, Edwards YJ, Ozdag H, Liu XZ, et al. Recurrent and private MYO15A mutations are associated with deafness in the Turkish population. Genet Test Mol Biomarkers. 2010;14(4):543–50.
Chang MY, Kim AR, Kim NK, Lee C, Lee KY, Jeon WS, Koo JW, Oh SH, Park WY, Kim D, et al. Identification and clinical implications of novel MYO15A mutations in a non-consanguineous Korean family by targeted exome sequencing. Mol Cells. 2015;38(9):781–8.
Nal N, Ahmed ZM, Erkal E, Alper OM, Luleci G, Dinc O, Waryah AM, Ain Q, Tasneem S, Husnain T, et al. Mutational spectrum of MYO15A: the large N-terminal extension of myosin XVA is required for hearing. Hum Mutat. 2007;28(10):1014–9.
Naz S, Imtiaz A, Mujtaba G, Maqsood A, Bashir R, Bukhari I, Khan MR, Ramzan M, Fatima A, Rehman AU, et al. Genetic causes of moderate to severe hearing loss point to modifiers. Clin Genet. 2017;91(4):589-98.
Belguith H, Aifa-Hmani M, Dhouib H, Said MB, Mosrati MA, Lahmar I, Moalla J, Charfeddine I, Driss N, Arab SB, et al. Screening of the DFNB3 locus: identification of three novel mutations of MYO15A associated with hearing loss and further suggestion for two distinctive genes on this locus. Genet Test Mol Biomarkers. 2009;13(1):147–51.
Brownstein Z, Friedman LM, Shahin H, Oron-Karni V, Kol N, Abu Rayyan A, Parzefall T, Lev D, Shalev S, Frydman M, et al. Targeted genomic capture and massively parallel sequencing to identify genes for hereditary hearing loss in middle eastern families. Genome Biol. 2011;12(9):R89.
Gao X, Zhu QY, Song YS, Wang GJ, Yuan YY, Xin F, Huang SS, Kang DY, Han MY, Guan LP, et al. Novel compound heterozygous mutations in the MYO15A gene in autosomal recessive hearing loss identified by whole-exome sequencing. J Transl Med. 2013;11:284.
Kalay E, Uzumcu A, Krieger E, Caylan R, Uyguner O, Ulubil-Emiroglu M, Erdol H, Kayserili H, Hafiz G, Baserer N, et al. MYO15A (DFNB3) mutations in Turkish hearing loss families and functional modeling of a novel motor domain mutation. Am J Med Genet A. 2007;143a(20):2382–9.
Liburd N, Ghosh M, Riazuddin S, Naz S, Khan S, Ahmed Z, Riazuddin S, Liang Y, Menon PS, Smith T, et al. Novel mutations of MYO15A associated with profound deafness in consanguineous families and moderately severe hearing loss in a patient with smith-Magenis syndrome. Hum Genet. 2001;109(5):535–41.
Shearer AE, Hildebrand MS, Webster JA, Kahrizi K, Meyer NC, Jalalvand K, Arzhanginy S, Kimberling WJ, Stephan D, Bahlo M, et al. Mutations in the first MyTH4 domain of MYO15A are a common cause of DFNB3 hearing loss. Laryngoscope. 2009;119(4):727–33.
Belyantseva IA, Boger ET, Friedman TB. Myosin XVa localizes to the tips of inner ear sensory cell stereocilia and is essential for staircase formation of the hair bundle. Proc Natl Acad Sci U S A. 2003;100(24):13958–63.
Liang Y, Wang A, Belyantseva IA, Anderson DW, Probst FJ, Barber TD, Miller W, Touchman JW, Jin L, Sullivan SL, et al. Characterization of the human and mouse unconventional myosin XV genes responsible for hereditary deafness DFNB3 and shaker 2. Genomics. 1999;61(3):243–58.
Fang Q, Indzhykulian AA, Mustapha M, Riordan GP, Dolan DF, Friedman TB, Belyantseva IA, Frolenkov GI, Camper SA, Bird JE. The 133-kDa N-terminal domain enables myosin 15 to maintain mechanotransducing stereocilia and is essential for hearing. Elife. 2015;4. https://doi.org/10.7554/eLife.08627.
King KA, Choi BY, Zalewski C, Madeo AC, Manichaikul A, Pryor SP, Ferruggiaro A, Eisenman D, Kim HJ, Niparko J, et al. SLC26A4 genotype, but not cochlear radiologic structure, is correlated with hearing loss in ears with an enlarged vestibular aqueduct. Laryngoscope. 2010;120(2):384–9.
Kim AR, Chung J, Kim NKD, Lee C, Park WY, Oh DY, Choi BY. The analysis of a frequent TMPRSS3 allele containing P.V116M and P.V291L in a Cis configuration among deaf Koreans. Int J Mol Sci. 2017;18(11).pii:E2246. https://doi.org/10.3390/ijms18112246.
Han KH, Oh DY, Lee S, Lee C, Han JH, Kim MY, Park HR, Park MK, NKD K, Lee J, et al. ATP1A3 mutations can cause progressive auditory neuropathy: a new gene of auditory synaptopathy. Sci Rep. 2017;7(1):16504.
Choi BY, Park G, Gim J, Kim AR, Kim BJ, Kim HS, Park JH, Park T, Oh SH, Han KH, et al. Diagnostic application of targeted resequencing for familial nonsyndromic hearing loss. PLoS One. 2013;8(8):e68692.
Kim BJ, Kim AR, Lee C, Kim SY, Kim NK, Chang MY, Rhee J, Park MH, Koo SK, Kim MY, et al. Discovery of CDH23 as a significant contributor to progressive Postlingual Sensorineural hearing loss in Koreans. PLoS One. 2016;11(10):e0165680.
Kim BJ, Kim AR, Han JH, Lee C, Oh DY, Choi BY. Discovery of MYH14 as an important and unique deafness gene causing prelingually severe autosomal dominant nonsyndromic hearing loss. J Gene Med. 2017;19(4). https://doi.org/10.1002/jgm.2950.
Park JH, Kim AR, Han JH, Kim SD, Kim SH, Koo JW, Oh SH, Choi BY. Outcome of Cochlear implantation in Prelingually deafened children according to molecular genetic etiology. Ear Hear. 2017;38(5):e316–24.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med: official J Am Coll Med Genet. 2015;17(5):405–24.
D'Aurizio R, Pippucci T, Tattini L, Giusti B, Pellegrini M, Magi A. Enhanced copy number variants detection from whole-exome sequencing data using EXCAVATOR2. Nucleic Acids Res. 2016;44(20):e154.
Svrakic M, Friedmann DR, Berman PM, Davis AJ, Roland JT Jr, Svirsky MA. Measurement of Cochlear implant electrode position from Intraoperative post-insertion skull radiographs: a validation study. Otol Neurotol : Am Otological Soc, Am Neurotol Soc Eur Acad Otol Neurotol. 2015;36(9):1486–91.
Chang MY, Kim AR, Kim NK, Lee C, Park WY, Choi BY. Refinement of molecular diagnostic protocol of auditory neuropathy Spectrum disorder: disclosure of significant level of etiologic homogeneity in Koreans and its clinical implications. Medicine. 2015;94(47):e1996.
Moteki H, Azaiez H, Sloan-Heggen CM, Booth K, Nishio SY, Wakui K, Yamaguchi T, Kolbe DL, Iwasa YI, Shearer AE, et al. Detection and confirmation of deafness-causing copy number variations in the STRC gene by massively parallel sequencing and comparative genomic hybridization. Ann Otol, Rhinol, laryngol. 2016;125(11):918–23.
Miyagawa M, Naito T, Nishio SY, Kamatani N, Usami S. Targeted exon sequencing successfully discovers rare causative genes and clarifies the molecular epidemiology of Japanese deafness patients. PLoS One. 2013;8(8):e71381.
Miyagawa M, Nishio SY, Hattori M, Moteki H, Kobayashi Y, Sato H, Watanabe T, Naito Y, Oshikawa C, Usami S. Mutations in the MYO15A gene are a significant cause of nonsyndromic hearing loss: massively parallel DNA sequencing-based analysis. Ann Otol, Rhinol, Laryngol. 2015;124(Suppl 1):158s–68s.
Rehman AU, Bird JE, Faridi R, Shahzad M, Shah S, Lee K, Khan SN, Imtiaz A, Ahmed ZM, Riazuddin S, et al. Mutational Spectrum of MYO15A and the molecular mechanisms of DFNB3 human deafness. Hum Mutat. 2016;37(10):991–1003.
Acknowledgements
None
Funding
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2015R1D1A1A01058225 to B.Y. Choi) and also by the Korean Health Technology R&D project, Ministry of Health & Welfare, Republic of Korea (HI15C1632 and HI14C1867) to B.Y. Choi).
Availability of data and materials
All data generated or analysed during this study are included in this published article and its Additional files.
Author information
Authors and Affiliations
Contributions
MYC analyzed data and wrote the manuscript. CL, JHH, MYK and HRP conducted the experiments and analyzed data. NK, WP and DYO analyzed data and contributed to writing the manuscript. BYC conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures in this study were approved by the Institutional Review Boards of Seoul National University Hospital (IRBY-H-0905-041-281) and Institutional Review Boards of Seoul National University Bundang Hospital (IRB-B-1007-105-402). Written informed consent was obtained from all participants.
Consent for publication
No individual patient data has been reported in the current study and consent for publication is not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. List of known 129 deafness genes targeted in this study. (DOCX 46 kb)
Additional file 2:
Table S2. Depth of coverage of customized panel sequencing. The table enumerates the mean coverage of the targeted regions from customized panel sequencing sample calculated by mpileup of samtools. (DOCX 206 kb)
Additional file 3:
Table S3. Exome sequencing statistics. The table lists information of human genome reference (v19), customized panel and whole exome sequencing aligned reads from each patient estimated by CalculateHsMetrics of picard tool. (DOCX 29 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chang, M.Y., Lee, C., Han, J.H. et al. Expansion of phenotypic spectrum of MYO15A pathogenic variants to include postlingual onset of progressive partial deafness. BMC Med Genet 19, 29 (2018). https://doi.org/10.1186/s12881-018-0541-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-018-0541-9