
Abstract
Background
Inactivating mutations of CDC73 cause Hyperparathyroidism-Jaw Tumour syndrome (HPT-JT), Familial Isolated Hyperparathyroidism (FIHP) and sporadic parathyroid carcinoma. We conducted CDC73 mutation analysis in an HPT-JT family and confirm carrier status of the proband’s daughter.
Methods
The proband had primary hyperparathyroidism (parathyroid carcinoma) and uterine leiomyomata. Her father and daughter had hyperparathyroidism (parathyroid adenoma) but no other manifestations of HPT-JT. CDC73 mutation analysis (sequencing of all 17 exons) and whole-genome copy number variation (CNV) analysis was done on leukocyte DNA of the three affecteds as well as the proband’s unaffected sister.
Results
A novel deletion of exons 4 to 10 of CDC73 was detected by CNV analysis in the three affecteds. A novel insertion in the 5’UTR (c.-4_-11insG) that co-segregated with the deletion was identified. By in vitro assay the 5’UTR insertion was shown to significantly impair the expression of the parafibromin protein. Screening for the mutated CDC73 confirmed carrier status in the proband’s daughter and the biochemistry and ultrasonography led to pre-emptive surgery and resolution of the hyperparathyroidism.
Conclusions
A novel gross deletion mutation in CDC73 was identified in a three-generation HPT-JT family emphasizing the importance of including screening for large deletions in the molecular diagnostic protocol.
Similar content being viewed by others

Background
Primary hyperparathyroidism (PHPT) is a common endocrine disorder affecting up to 2% of individuals over the age of 55 years [1]. It is caused by solitary benign adenoma in 80–85%, hyperplasia in 10–15%, and parathyroid carcinoma in less than 1%. In up to 10% of cases PHPT is part of a familial syndrome such as multiple endocrine neoplasia (MEN) types 1 or 2, hyperparathyroidism-jaw tumor syndrome (HPT-JT), familial isolated hyperparathyroidism (FIHP) or familial hypocalciuric hypercalcemia [2]. In the HPT-JT syndrome, carcinomas account for approximately 15% of the parathyroid tumors [3].
The most common manifestations of the autosomal dominant HPT-JT syndrome are parathyroid tumours and ossifying fibromas of the maxilla and mandible. Patients may also develop renal abnormalities and uterine tumors [4]. The HPT-JT syndrome is caused by mutations of the cell division cycle protein 73 homolog (CDC73) gene, at chromosome 1q31.2 [5]. The 17 exons of the CDC73 tumor suppressor gene encode the predominantly nuclear, 531-amino acid protein, parafibromin [6]. Parafibromin regulates gene transcription as part of the RNA polymerase II-associated polymerase-associated factor 1 (PAF1) complex that has a fundamental role in chromatin remodelling [6, 7]. Parafibromin inhibits cell proliferation by blocking the expression of cyclin D1 [8] and as a component of the Wnt signalling pathway [9]. Moreover, the CDC73 gene is implicated in sporadic parathyroid carcinomas and mutations are present in up to 70% of cases [10, 11]. Parafibromin can serve as a parathyroid carcinoma marker. While it is expressed in normal parathyroid glands, parathyroid adenoma and hyperplasia, it is usually absent in parathyroid carcinomas and some atypical adenomas [12].
The majority of the CDC73 gene loss-of-function mutations associated with hyperparathyroidism and parathyroid carcinoma are frame-shift, nonsense or missense occurring within the protein-encoding exons [13]. Recently, some HPT-JT and FIHP cases without these types of mutation were shown to have intragenic or whole deletion of CDC73 [14,15,16,17,18,19].
We report here a 3-generation HPT-JT syndrome family in which although initial analysis identified a variant in the 5’UTR of the mRNA no mutation was found in the protein-coding exons or exon/intron junctions. Further analysis revealed an intragenic deletion of exons 4–10 of CDC73 that co-segregated with the 5’UTR variant in the affected individuals.
Methods
Patients
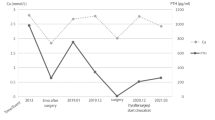
The proband (III-1, Fig. 1) was a 48-year-old woman who presented for evaluation of weight loss and fatigue. She had a history of nausea, polydipsia and polyuria but not of constipation or nephrolithiasis. Physical examination was remarkable for a cachectic appearance (BMI = 18.7 kg/m2) and the presence of a neck mass in the region of the right superior thyroid lobe that was hard on palpation. Laboratory tests revealed markedly elevated total serum calcium of 4.21 mmol/L (normal ≤2.60) and serum parathyroid hormone (PTH) of 133 pmol/L (normal <7.6). The patient’s hypercalcemia was emergently treated with intravenous fluids, furosemide and pamidronate.

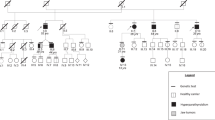
Pedigree of family with HPT-JT and CDC73 gene mutation. Clinical status is indicated by open symbols (unaffected or status not known) and solid symbols (affected). Proband is indicated by the arrow. The presence (+) or absence (−) of the 5’UTR variant/exon 4–10 deletion in tested family members is shown
Ultrasound imaging and sestamibi scan suggested the presence of ~3 cm neck mass in close proximity to the posterior aspect of the right hemithyroid lobe. Parathyroid exploration revealed a local gross invasion of the mass into surrounding structures, and a right hemithyroidectomy in addition to parathyroidectomy was performed. Pathological evaluation of the 3.5 cm surgical specimen (Fig. 2a and b) revealed vascular invasion (Fig. 2c), thyroid invasion (Fig. 2d) and nuclear atypia (Fig. 2e). Immunochemistry demonstrated PTH positivity and absence of thyroglobulin staining. A diagnosis of parathyroid carcinoma was made, and while there were no metastatic lymph nodes, the margins were positive. After surgery, the patient developed reactive hypocalcemia due to hungry bone syndrome, which was managed with oral calcium supplementation. After initial discharge she was readmitted on two occasions for marked hypocalcemia requiring intravenous or oral calcium and calcitriol. This issue had resolved by the time she received radiation therapy 3 months after surgery, but she developed radiation-induced hypothyroidism requiring oral levothyroxine replacement. Her weight improved and she is asymptomatic with no evidence of disease recurrence 6 years post-treatment.

Gross view (a) and histology (b-e) of parathyroid carcinoma surgical specimen from proband (III-1, Fig. 1). a Tumor with features of fibrous banding. b Tumor overview. c Vascular invasion. d Thyroid invasion. e Nuclear atypia
A search for other manifestations of the HPT-JT syndrome was conducted. Dental panoramic radiograph, bone scan, renal and hepatic ultrasounds were normal. However, approximately 18 months after initial diagnosis a uterine leiomyoma [20] of 6.3 by 4.8 by 4.5 cm was found on ultrasound during work-up for menorrhagia and was treated surgically.
The patient’s first-degree family members were also evaluated. The father (II-2, Fig. 1) had a 2.2 cm parathyroid adenoma removed at the age of 32, but other manifestations of HPT-JT were not noted. The daughter (IV-1, Fig. 1) underwent screening biochemistry and was found to have PHPT (total serum calcium, 3.1 mmol/L, PTH, 8.7 pmol/L). Ultrasound imaging demonstrated a hypoechoic nodule posterior to the left hemithyroid lobe, and she underwent surgical excision of a parathyroid adenoma.
Genetic testing
The Institutional Research Ethics Board of the IRCCS Casa Sollievo della Sofferenza Hospital approved the protocol and informed consent was obtained from the proband and family members. Genomic DNA was extracted from peripheral white blood cells using standard methods. The entire coding sequence of the CDC73 gene including the exon-intron boundaries was sequenced by PCR amplification and direct sequencing of all the 17 exons (16 amplicons) as previously described [12]. Moreover DNA extracted from formalin fixed paraffin embedded (FFPE) tumour tissue excised from the affected daughter was screened and loss of heterozygosity (LOH) investigated as described previously [12]. No other germline DNA or FFPE tissue was available at the time of the study.
pGL3 constructs and luciferase assay
The 184 bp 5’UTR sequence of the mRNA (encoded by the CDC73 gene) was PCR amplified from normal human DNA with the forward primer having a HindIII and the reverse primer having a NcoI restriction site. The PCR product, after digestion with these enzymes (New England Biolabs), was cloned into the HindIII/NcoI digested pGL3 Basic vector (Promega). The NcoI site was removed and the Kozak sequence restored by specific mutagenesis to obtain the WT-5’UTR-pGL3 construct. By site-directed mutagenesis with this construct as template an additional G was introduced into the polyG (n = 8) tract terminating 3 bases upstream of the ATG start site to obtain the MUT-5’UTR-pGL3 construct. Sequences of mutagenesis primers and methods are provided in the Additional file 1. Correctness of the constructs was confirmed by sequencing.
Human embryonic kidney (HEK293) cells were seeded in 6 well plates in DMEM-F12 supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Twenty-four hours later cells were transfected (Lipofectamine 2000 – Invitrogen) with increasing concentrations of pGL3 Basic, WT-5’UTR-pGL3 or MUT-5’UTR-pGL3. After 48 h, medium was removed, cells were washed, and lysed, before being scraped from the wells and collected. After vortexing for 15 s and centrifugation at 12000 rpm (2 min at 4 °C), aliquots of the supernatant were added to Luciferase Assay reagent (Promega) and the luciferase activity read in a luminometer.
5' UTR-parafibromin expression constructs and western blot
The 184 bp 5’UTR sequence of the mRNA (encoded by the CDC73 gene) was PCR amplified from normal human DNA with the forward primer having an EcoRI and the reverse primer having an SgfI restriction site. The PCR product, after digestion with these enzymes, was cloned (upstream of the open reading frame) into the EcoRI/SgfI digested pCMV6 vector that expresses the human CDC73 cDNA encoding parafibromin Myc/Flag-tagged at its COOH-terminus (Origene RC209479) [21].
The SgfI site was deleted and the 5′ UTR sequence restored by mutagenesis to generate the WT-5’UTR-Flag construct. By site-directed mutagenesis with this construct as template an additional G was introduced into the polyG (n = 8) tract terminating 3 bases upstream of the ATG start site to obtain the MUT-5’UTR-Flag construct. Sequences of mutagenesis primers and methods are provided in the Additional file 1. Correctness of the constructs was confirmed by sequencing.
HEK293 cells were cultured and transfected as described above with the WT-5’UTR-Flag and MUT-5’UTR-Flag constructs. Total cellular proteins were extracted in radioimmunoprecipitation (RIPA buffer) (150 mM NaCl, 50 mM Tris-HCl, 1% Nonidet P-40, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate, pH 8.0) supplemented with Complete EDTA-Free Protease Cocktail Inhibitor (1 tablet/10 mL RIPA buffer). Protein aliquots were electrophoresed through 8% SDS polyacrylamide gels, electrotransferred to PVDF membrane (Millipore, Billerica, MA), blotted overnight at 4 °C with rabbit anti-Flag monoclonal antibody (Cell Signaling Technology) and for 1 h at room temperature with horseradish peroxidase-conjugated goat anti-rabbit IgG antibody (Biorad). Membranes were stripped and blotted with β-tubulin rabbit monoclonal antibody (Cell Signaling Technology). Densitometric analysis was made with ImageJ (http://rsbweb.nih.gov/ij/).
SNP array analysis
Whole-genome copy number variation (CNV) analysis was carried out with the CytoScan HD array platform (Affymetrix, Santa Clara, CA) on leukocyte DNA of the proband (III-1, Fig. 1), her father II-2, her daughter IV-1 and her sister III-3. The array contains more than 2.6 million markers for copy number analysis and approximately 750,000 SNPs that fully genotype with greater than 99% accuracy. The CytoScanHD assay was performed according to the manufacturer’s protocol, starting with 250 ng DNA. DNA was digested with the NspI restriction enzyme, ligated to an appropriate adapter for the enzyme, and subjected to PCR amplification using a single primer. After digestion with DNase I, the PCR products were labeled with a biotinylated nucleotide analogue, using terminal deoxynucleotidyl transferase. Hybridization to the microarray was carried out in a Hybridization Oven 645 while subsequent washing and staining were performed using the Fluidics Station 450. The array was then scanned with the Scanner 3000 7G and both the quality control step and copy number analysis were performed using Chromosome Analysis Suite Software version 2.0. The raw data file (.CEL) was normalized using the default options and an unpaired analysis was performed using as baseline 270 HapMap samples to obtain the copy number value from. CEL files while the amplified and/or deleted regions were detected using a standard Hidden Markov Model (HMM) method. Base pair positions were obtained from the University of California Santa Cruz (UCSC) Genome Browser (http://genome-euro.ucsc.edu/cgi-bin/hgGateway?redirect=manual&source=genome.ucsc.edu), build GRCh37 (hg19).
Statistics
Data are expressed as mean ± SE of triplicate estimations with each experiment repeated three times and a p value <0.05 was considered statistically significant.
Results
CDC73 mutation screening
CDC73 sequence analysis of genomic DNA of whole blood of the proband (III-I, Fig. 1) did not reveal a mutation in the coding region or at splice sites. However, within the 5’UTR an insertion of an additional guanidine within a well-conserved polyG tract (n-8), 3 bp upstream of the ATG start site, namely c.-4_-11insG, was found (Figs. 3a and b). In addition, the SNP array analysis revealed an interstitial microdeletion of 0.25 Mb in band 1q31.2 (Fig. 4) in cis with the 5’UTR variant. The hemizygous region encompassed part of the CDC73 coding sequence from exon 4 to 10 (Fig. 4). These same changes (5′ UTR variant, gene deletion) were identified in genomic DNA of the affected father (II-2, Fig. 1) and the affected daughter (IV-1, Fig. 1) of the proband. These changes were not found in genomic DNA of the unaffected sister (III-3, Fig. 1) of the proband. Sequencing and LOH analysis of FFPE parathyroid tissue of the proband (III-1, Fig. 1) was uninformative with respect to identifying a somatic second hit involving the wild-type CDC73 allele.

a. Species sequence alignment of CDC73 encoding the proximal 5’UTR. The g tract (n = 8) and the ATG start codon are boxed. b Sequence chromatogram of leukocyte genomic DNA of proband (III-1, Fig. 1) showing heterozygosity for insertion of an additional guanidine in the tract of eight guanidines (c.–4_11insG). c Luciferase activity (mean ± SE) of cells transfected with either WT-5’UTR-pGL3 or MUT-5’UTR-pGL3 constructs. See text for details. *, p < 0.05. d (i) Parafibromin (Flag) and β-tubulin (Tubulin) western blots of cells transfected with either WT-5’UTR-Flag or MUT-5’UTR-Flag constructs. (ii) Densitometric analysis. **, p < 0.01. See text for details

CytoScan HD Array analysis results of the patient. Intensity data (log 2 ratio value) of each probe is drawn along chromosome 1 from 193.00 to 193.22 Mb (USCS Genome Browser build February 2009, hg19). The red bar represents the 1q31.2 deletion identified, encompassing exons 4 to 10 of the CDC73 gene
In vitro functional analysis of the 5’UTR variant
In the first approach, HEK293 cells were transfected with the pGL3 Basic construct in which the CDC73 5’UTR (either wildtype or mutant) had been inserted upstream of the luciferase coding sequence. While at low concentration (50 ng) of DNA transfected luciferase activity was minimal (no different from mock-transfected cells, data not shown), with higher concentrations of DNA (250 ng) significant luciferase activity was noted with the WT-5’UTR-pGL3 construct whereas the activity of the MUT-5’UTR-pGL3 construct was significantly less (Fig. 3c).
In the second approach, HEK293 cells were transfected with an expression vector in which the wild type or mutant CDC73 5’UTR had been inserted upstream of a parafibromin cDNA having a Flag epitope encoded at its COOH-terminus. Western blot analysis of cell protein extracts revealed markedly reduced levels of the exogenous parafibromin in cells transfected with the MUT-5’UTR-Flag construct as compared with those transfected with the WT-5’UTR-Flag construct [Fig. 3d (i) and (ii)].
Discussion
Here we describe a rare case of HPT-JT syndrome, in which the hyperparathyroidism in affected family members co-segregated with an altered CDC73 allele harboring a large intragenic deletion and a 5’UTR variant. While deletion of exons CDC73 4–10 is clearly pathogenic, our in vitro analysis also suggests impaired function of the CDC73 mRNA having the c.-4_-11insG alteration However, species sequence comparison (UCSC, https://genome.ucsc.edu/) reveals that the 8G–tract shows limited phylogenetic conservation from humans to rodents, and in lower species the length and sequence vary (Fig 3a). With respect to the 5’UTR variant, recently, it appeared in the ClinVar database (https://www.ncbi.nlm.nih.gov/projects/SNP/, http://www.internationalgenome.org/ and https://www.ncbi.nlm.nih.gov/clinvar/, code rs886043365 and 286,328, respectively), but it was not found in ExAC or gnomAD, (http://exac.broadinstitute.org/, http://gnomad.broadinstitute.org/) databases. In the ClinVar database, Minor Allele Frequency (MAF) was not available. Mutations were identified only 5 times in subjects affected by HPT-JT (2 cases), parathyroid carcinoma (1 case), and in 2 cases with an unreported clinical condition. The reported clinical significance was not unanimous, being “uncertain” in 3 cases and “benign or likely benign” in 2 cases.
In our family case, in which the 5’UTR variant is in cis with the large deletion, we believe that affected status was due to the loss of genomic sequence, without any ascribable influence of the 5’UTR variant. Further ad hoc studies and genetic testing on larger cohorts would help to clarify this issue.
Mutations of the CDC73 gene have presented as missense/nonsense, frameshift insertion or deletion that are scattered throughout the entire coding sequence with selectivity for some exons as opposed to others [12, 13]. As such mutations have not been found in all HPT-JT syndrome cases and its variants, the search for large genomic deletions at the CD73C locus has recently intensified, leading to their identification in several cases [14,15,16,17,18,19] (see Table 1). In one large study [16], large gene deletions represented 35% of all the CDC73 genetic lesions identified, regardless of phenotypic presentation (sporadic parathyroid carcinoma, FIHP or HPT-JT), suggesting a possible underestimation of the presence of such genomic rearrangements at the CDC73 locus in the pathogenicity of the syndrome.
Identification of a somatic mutation in the wild-type CDC73 allele in the tumor has been important in confirming that the “second hit” hypothesis for tumor suppressor genes applies to CDC73 (e.g., [22]). In the present case, sequencing and LOH analysis were unsuccessful in identifying a somatic second hit. In such cases in which absent parafibromin immunostaining in the tumor has been documented, a variety of mechanisms have been proposed whereby the parafibromin expression could be reduced. These include gene inactivation by hypermethylation of the CDC73 promoter, an event that appears to be rare or absent in HPT-JT tumors [23, 24]. Evidence of inhibition of the CDC73 gene by a transcription factor [25] or by a microRNA suppressing the CDC73 mRNA [26] has been provided in squamous cell carcinoma but has yet to be confirmed for parathyroid neoplasia.
The present study has important implications for the clinical management of parathyroid carcinoma as occurring sporadically or within families [27, 28]. Patients presenting with apparently sporadic parathyroid carcinomas may carry germline mutations in the CDC73 gene, and may thus have the potential to express the HPT-JT syndrome or a variant. Family members may also be mutation carriers. CDC73 genetic and clinical testing should be considered for the following: patients with a suspicion or diagnosis of parathyroid carcinoma; family members of patients with a diagnosis of parathyroid carcinoma; and patients with a positive family history of parathyroid tumor of any sort. For asymptomatic individuals (e.g., family members) the following is recommended: periodic biochemical screen (serum total calcium and PTH): tumor surveillance, e.g., panoramic jaw x-ray, kidney and uterine ultrasound. For family members of patients with parathyroid carcinoma periodic biochemical screen regardless of CDC73 status is recommended.
Conclusions
The presence of a CDC73 mutation is associated with increased risk of parathyroid carcinoma. Nevertheless, as emphasized here and in other reports, the absence of pathogenic coding variants does not exclude large genomic deletion, the search for which should be encouraged. If known preoperatively, this information will be helpful to the surgeon in planning the extent of surgery required. As the risk of recurrence has to be weighed against the possibility of incomplete disease penetrance, the optimal extent of parathyroid surgery for benign disease in the setting of a CDC73 mutation remains challenging and controversial [29,30,31,32,33].
Change history
13 September 2017
An erratum to this article has been published.
Abbreviations
- BMI:
-
Body Mass Index
- CNV:
-
Copy Number Variations
- FFPE:
-
Formalin Fixed Paraffin Embedded
- FIHP:
-
Familial Isolated Hyperparathyroidism
- HEK293:
-
Human Embryonic Kidney 293
- HMM:
-
Hidden Markov Model
- HPT-JT:
-
Hyperparathyroidism with Jaw Tumour
- MEN1:
-
Multiple Endocrine Neoplasia 1
- PAF1:
-
Polymerase Associated Factor 1
- PHPT:
-
Primary Hyperparathyroidism
- UTR:
-
Untranslated Region
References
DeLellis RA, Mazzalia P, Mangray S. Primary hyperparathyroidism: a current perspective. Arch Pathol Lab Med. 2008;1251:62.
Hendy GN, Cole DEC. Genetic defects associated with familial and sporadic hyperparathyroidism. Front Horm Res. 2013;41:149–65.
DeLellis RA. Challenging lesions in the differential diagnosis of endocrine tumors: parathyroid carcinoma. Endocr Pathol. 2008;19:221–5.
Chen JD, Morrison C, Zhang C, Kahnoski K, Carpten JD, Teh BT. Hyperparathyroidism-jaw tumor syndrome. J Intern Med. 2003;253:634–42.
Carpten JD, Robbins CM, Villablanca A, Forsberg L, Presciuttini S, Bailey-Wilson J, Simonds WF, Gillanders EM, Kennedy AM, Chen JD, Agarwal SK, Sood R, Jones MP, Moses TY, Haven C, Petillo D, Leotlela PD, Harding B, Cameron D, Pannett AA, Höög A, Heath H 3rd, James-Newton LA, Robinson B, Zarbo RJ, Cavaco BM, Wassif W, Perrier ND, Rosen IB, Kristoffersson U, Turnpenny PD, Farnebo LO, Besser GM, Jackson CE, Morreau H, Trent JM, Thakker RV, Marx SJ, Teh BT, Larsson C, Hobbs MR. HRPT2 encoding parafibromin is mutated in hyperparathyroidism–jaw tumor syndrome. Nat Gen. 2002;32:676–80.
Yart A, Gstaiger M, Wirbelauer C, Pecnik M, Anastasiou D, Hess D, Krek W. The HRPT2 tumor suppressor gene product parafibromin associates with human PAF1 and RNA polymerase II. Mol Cell Biol. 2005;25:5052–60.
Rozenblatt-Rosen O, Hughes CM, Nannepaga SJ, Shanmugam KS, Copeland TD, Guszczynski T, Resau JH, Meyerson M. The parafibromin tumor suppressor protein is part of a human Paf1 complex. Mol Cell Biol. 2005;25:612–20.
Woodard GE, Lin L, Zhang J-H, Agarwal SK, Marx SJ, Simonds WF. Parafibromin, product of the hyperparathyroidism-jaw tumor syndrome gene HRPT2, regulates cyclinD1/PRAD1 expression. Oncogene. 2005;24:1272–5.
Mosimann C, Hausmann G, Basler K. Parafibromin/hyrax activates Wnt/Wg target gene transcription bydirect association with beta-catenin/armadillo. Cell. 2006;125:327–41.
Howell VM, Haven CJ, Kahnoski K, Khoo SK, Petillo D, Chen J, Fleuren GJ, Robinson BG, Delbridge LW, Philips J, Nelson AE, Krause U, Hammje K, Dralle H, Hoang-Vu C, Gimm O, Marsh DJ, Morreau H, Teh BT. HRPT2 mutations are associated with malignancy in sporadic parathyroid tumors. J Med Gen. 2003;40:657–3.
Shattuck TM, Välimäki S, Obara T, Gaz RD, Clark OH, Shoback D, Wierman ME, Tojo K, Robbins CM, Carpten JD, Farnebo LO, Larsson C, Arnold A. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med. 2003;349:1722–9.
Guarnieri V, Battista C, Muscarella LA, Bisceglia M, de Martino D, Baorda F, Maiello E, D'Agruma L, Chiodini I, Clemente C, Minisola S, Romagnoli E, Corbetta S, Viti R, Eller-Vainicher C, Spada A, Iacobellis M, Malavolta N, Carella M, Canaff L, Hendy GN, Cole DE, Scillitani A. CDC73 mutations and parafibromin immunhistochemistry in parathyroid tumors: clinical correlations in a single-centre patient cohort. Cell Oncol (Dordrecht). 2012;35:411–22.
Newey PJ, Bowl MR, Cranston T, Thakker RV. Cell division cycle protein 73 homolog (CDC73) mutations in the hyperparathyroidism-jaw tumor syndrome (HPT-JT) and parathyroid tumors. Hum Mut. 2010;31:295–307.
Cascón A, Huarte-Mendicoa CV, Javier Leandro-García L, Letón R, Suela J, Santana A, Costa MB, Comino-Méndez I, Landa I, Sánchez L, Rodríguez-Antona C, Cigudosa JC, Robledo M. Detection of the first gross CDC73 germline deletion in an HPT-JT syndrome family. Genes Chromosomes Cancer. 2011;50:922–9.
Domingues R, Tomaz RA, Martins C, Nunes C, Bugalho MJ, Cavaco BM. Identification of the first germline HRPT2 whole-gene deletion in a patient with primary hyperparathyroidism. Clin Endocrinol. 2012;76:33–8.
Bricaire L, Odou MF, Cardot-Bauters C, Delemer B, North MO, Salenave S, Vezzosi D, Kuhn JM, Murat A, Caron P, Sadoul JL, Silve C, Chanson P, Barlier A, Clauser E, Porchet N. Groussin L; GTE group. Frequent large germline HRPT2 deletions in a French national cohort of patients with primary hyperparathyroidism. J Clin Endocrinol Metab. 2013;98:E403–8.
Korpi-Hyövälti E, Cranston T, Ryhänen E, Arola J, Aittomäki K, Sane T, Thakker RV, Schalin-Jäntti C. CDC73 intragenic deletion in familial primary hyperparathyroidism associated with parathyroid carcinoma. J Clin Endocrinol Metab. 2014;99:3044–8.
Kong J, Wang O, Nie M, Shi J, Hu Y, Jiang Y, Li M, Xia W, Meng X, Xing X. Familial isolated primary hyperparathyroidism/hyperparathyroidism-jaw tumour syndrome caused by germline gross deletion or point mutations of CDC73 gene in Chinese. Clin Endocrinol. 2014;81:222–30.
Davidson JT, Lam CG, McGee RB, Bahrami A, Diaz-Thomas A. Parathyroid cancer in the pediatric patient. J Pediatr Hematol Oncol. 2016;38:32–7.
Bradley KJ, Hobbs MR, Buley ID, Carpten JD, Cavaco BM, Fares JE, Laidler P, Manek S, Robbins CM, Salti IS, Thompson NW, Jackson CE, Thakker RV. Uterine tumours are a phenotypic manifestation of the hyperparathyroidism-jaw tumour syndrome. J Intern Med. 2005;257:18–26.
Pazienza V, la Torre A, Baorda F, Alfarano M, Chetta M, Muscarella LA, Battista C, Copetti M, Kotzot D, Kapelari K, Al-Abdulrazzaq D, Perlman K, Sochett E, Cole DE, Pellegrini F, Canaff L, Hendy GN, D'Agruma L, Zelante L, Carella M, Scillitani A, Guarnieri V. Identification and functional characterization of three NoLS (nucleolar localisation signals) mutations of the CDC73 gene. PLoS One. 2013;8:e82292.
Bradley KJ, Cavaco BM, Bowl MR, Harding B, Cranston T, Fratter C, Besser GM, Conceição Pereira M, Davie MW, Dudley N, Leite V, Sadler GP, Seller A, Thakker RV. Parafibromin mutations in hereditary hyperparathyroidism syndromes and parathyroid tumors. Clin Endocrinol. 2006;64:299–306.
Hewitt KM, Sharma PK, Samowitz W, Hobbs M. Aberrant methylation of the HRPT2 gene in parathyroid carcinoma. Ann Otol Rhinol Laryngol. 2007;116:928–33.
Hahn MA, Howell VM, Gill AJ, Clarkson A, Weaire-Buchanan G, Robinson BG, Delbridge L, Gimm O, Schmitt WD, Teh BT, Marsh DJ. CDC73/HRPT2 CpG island hypermethylation and mutation of 5′-untranslated sequence are uncommon mechanisms of silencing parafibromin in parathyroid tumors. Endocr Relat Cancer. 2010;18:273–82.
Rather MI, Swamy S, Gopinath KS, Kumar A. Transcriptional repression of tumor suppressor CDC73, encoding an RNA polymerase II inhibitor, by Wilms tumor 1 protein (WT1) promotes cell proliferation. Implication for cancer therapeutics. J Biol Chem. 2014;289:968–76.
Rather MI, Nagashri MN, Swamy SS, Gopinath KS, Kumar A. Oncogenic microRNA-155 downregulates tumor suppressor CDC73 and promotes oral squamous cell carcinoma cell proliferation. Implications for cancer therapeutics. J Biol Chem. 2013;288:608–18.
Guarnieri V, Scillitani A, Muscarella LA, Battista C, Bonfitto N, Bisceglia M, Minisola S, Mascia ML, D'Agruma L, Cole DE. Diagnosis of parathyroid tumors in familial isolated hyperparathyroidism with HRPT2 mutation: implications for cancer surveillance. J Clin Endocrinol Metab. 2006;91:2827–32.
Kelly TG, Shattuck TM, Reyes-Mugica M, Stewart AF, Simonds WF, Udelsman R, Arnold A, Carpenter TO. Surveillance for early detection of aggressive parathyroid disease: carcinoma and atypical adenoma in familial isolated hyperparathyroidism associated with a germline HRPT2 mutation. J Bone Miner Res. 2006;21:1666–71.
Sarquis MS, Silveira LG, Pimenta FJ, Dias EP, Teh BT, Friedman E, Gomez RS, Tavares GC, Eng C, De Marco L. Familial hyperparathyroidism: surgical outcome after 30 years of follow-up in three families with germline HRPT2 mutations. Surgery. 2008;143:630–4.
Silveira LG, Dias EP, Marinho BC, Gomez RS, De Marco L, Sarquis MS. HRPT2-related familial isolated hyperparathyroidism: could molecular studies direct the surgical approach? Arq Bras Endocrinol Metabol. 2008;143:1211–20.
Guarnieri V, Bisceglia M, Bonfitto N, Cetani F, Marcocci C, Minisola S, Battista C, Chiodini I, Cole DE, Scillitani A. Re: familial hyperparathyroidism: surgical outcome after 30 years of follow-up in three families with germline HRPT2 mutations. Surgery. 2008;144:839–40.
Iacobone M, Barzon L, Porzionato A, Masi G, Macchi V, Viel G, Favia G. The extent of parathyroidectomy for HRPT2-related hyperparathyroidism. Surgery. 2009;145:250–1.
Mehta A, Patel D, Rosenberg A, Baufragech M, Ellis RJ, Nilubol N, Quezado MM, Marx SJ, Simonds WF, Kebebew E. Hyperparathyroidism-jaw tumor syndrome: results of operative management. Surgery. 2014;156:1315–24.
Acknowledgements
We thank all family members for their participation. We gratefully acknowledge Angelo Sparaneo for helpful assistance.
Funding
This research was supported by the Ricerca Corrente and GR Familia Project 2011–02351489 funding granted by the Italian Ministry of Health (to VG) and by the “5 × 1000” voluntary contributions and by a Canadian Institutes of Health Research Operating Grant (to GNH). The funders had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
The datasets during and/or analysed during the current study available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
FB and AYS performed molecular screening; OP performed array CGH; RMS, CK, MJD and SR followed clinically the family; AS, GNH and DECC contributed to critical revision of the work and revised/edited the manuscript. VG, GNH and DEC conceived the work and wrote the manuscript. All the Authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent for molecular analysis was obtained from the patient or in the case of children, from their parent or legal guardian. A copy of the consent form is available. The Institutional Research Ethics Board of the IRCCS Casa Sollievo della Sofferenza Hospital approved the protocol (Familia-ProtN11/CE-Jan 16).
Consent for publication
Consent to publish data, images or videos was obtained from any subject, or in the case of children, from their parent or legal guardian.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional information
The original version of this article was revised: Minor mistakes were corrected in the table on page 6 and in the Abbreviations section on page 7.
An erratum to this article is available at https://doi.org/10.1186/s12881-017-0459-7.
Additional file
Additional file 1:
The file contains the sequences of the mutagenesis primers and the mutagenesis protocol. (DOCX 12 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Guarnieri, V., Seaberg, R.M., Kelly, C. et al. Large intragenic deletion of CDC73 (exons 4–10) in a three-generation hyperparathyroidism-jaw tumor (HPT-JT) syndrome family. BMC Med Genet 18, 83 (2017). https://doi.org/10.1186/s12881-017-0445-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-017-0445-0