
Abstract
Background
Cataract is a major cause of severe visual impairment in childhood. The purpose of this study was to determine the genetic cause of syndromic congenital cataract in an Australian mother and son.
Method
Fifty-one genes associated with congenital cataract were sequenced in the proband using a custom Ampliseq library on the Ion Torrent Personal Genome Machine (PGM). Reads were aligned against the human genome (hg19) and variants were annotated. Variants were prioritised for validation by Sanger sequencing if they were novel, rare or previously reported to be associated with paediatric cataract and were predicted to be protein changing. Variants were assessed for segregation with the phenotype in the affected mother.
Result
A novel likely pathogenic variant was identified in the transactivation domain of the MAF gene (c.176C > G, p.(Pro59Arg)) in the proband and his affected mother., but was absent in 326 unrelated controls and absent from public variant databases.
Conclusion
The MAF variant is the likely cause of the congenital cataract, Asperger syndrome, seizures, hearing loss and facial characteristics in the proband, providinga diagnosis of Aymé-Gripp syndrome for the family.
Similar content being viewed by others


Background
Cataract is an opacity of the crystalline lens resulting in impaired vision. Cataract formation is typically an age-related process that affects adults; however, rarely it can be present at birth or early childhood and is classified as congenital (or juvenile/paediatric) cataract. Congenital cataract occurs in 1–6 per 10,000 live births in developed countries [1]; in Australia the incidence is estimated to be 2.2 per 10,000 births [2].
Around 50% of cases have a genetic cause [3], with other causes including intrauterine infection, malnutrition and metabolic disorder. Hereditary congenital cataracts can be transmitted as autosomal recessive, autosomal dominant or X-linked traits, with autosomal dominant the most common mode of inheritance, and can be isolated or syndromic (associated with additional non-ocular abnormalities) [4]. The disorder demonstrates genetic and phenotypic heterogeneity.
Among the many genes associated with congenital cataracts is the transcription factor gene MAF (v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog, OMIM 177075, NM_005360.4). The MAF family of transcription factors is divided into two subgroups, large and small. The large subgroup (MAFA, MAFB, c-MAF or v-MAF, and retina-specific leucine zipper (NRL)) is characterized by a bZip structure, a motif for DNA binding and protein dimerization and a transactivation domain [5]. The small MAF proteins (MAFF, MAFG, and MAFK) lack the transactivation domain [5, 6].
Here we report a missense variant in v-MAF, usually referred to as MAF, in a mother and son with syndromic congenital cataract associated with hearing loss and developmental delay.
Methods
DNA was extracted from whole blood using the QIAamp DNA blood maxi kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Fifty-one genes associated with congenital cataract human or mouse were selected through a review of the literature (Additional file 1: Table S1) [7,8,9,10,11,12,13,14,15,16,17,18,19,20] for sequencing the coding and untranslated regions.
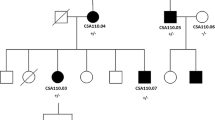
A sequencing library was prepared using the proband’s DNA (patient CSA108.01, Fig. 1a) as template. The library was generated with the Ion AmpliSeq library kit version 2.0 (Life Technologies, California, USA) and custom Ion Ampliseq primers according to the manufacturer’s protocols, and was sequenced on an Ion Torrent Personal Genome Machine using the Ion PGM Sequencing 200 Kit v2 and an Ion 318 chip (Life Technologies). Alignment to the reference genome (hg19), variant calling and annotation were conducted in Torrent Suite (version 3.6.), and Ion reporter (V4.0) with appropriate plugins. Variants were prioritized for further analysis if they were predicted to alter protein sequence (non-synonymous), were absent or very rare (Minor Allele Frequency < 1%) in public databases including dbSNP137 (https://www.ncbi.nlm.nih.gov/SNP/), Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org/) and absent from an in-house list of sequencing errors previously seen on this gene panel. In addition, selected variants were assessed by SIFT [21] and Polyphen-2 [22] for their predicted effect on protein function.

a Pedigree of family CSA108 with variant in MAF. Individuals with ID numbers were examined by an ophthalmologist. Solid circles indicate affected females and solid squares indicate affected males. The proband is marked by an arrow head. “+” indicates mutant allele and “−” indicates wild type allele of c.176C > G in the MAF gene. b Sequence chromatogram of two examined individuals at variant c.176C > G. Both sequenced affected members are heterozygous for this variant. c Protein alignment shows the MAF protein is highly conserved among the indicated species. The mutated residue is indicated by the box
The detected novel, coding variant in MAF was validated by Sanger sequencing using forward primer 5′-GGGGGTGTGTGTGTGAGC-3′ and reverse primer 5′-CTGGAGCTGGTGGCTGTT-3′. PCR reactions of 20 μl final volume consisting of 1X Coraload PCR buffer (Qiagen), 0.1 mM dNTPs (Roche Diagnostics, Basel, Switzerland), 0.5 μM each primer, 0.5U Hot Star Plus Taq Polymerase (Qiagen) and 40 ng of DNA was prepared. Final concentrating of Mg2+ was adjusted to 2.5 mM by adding the required amount of Mgcl2 (Qiagen). Amplification conditions involved an initial activation step of 95 °C for 5 min, followed by 35 cycles of 30 s of denaturation at 95 °C, 30 s of annealing at 57 °C and 30 s of extension at 72 °C. A final extension step was for 5 min at 72 °C. The PCR products were cleaned by treatment with 10 units (U) Exonuclease I (New England Biolabs, Genesearch Pty Ltd, QLD, Australia) and 2 U of Shrimp Alkaline Phosphatase (SAP) (USB, Millennium Science Pty. Ltd., VIC, Australia) at 37 °C for 1 h followed by enzyme deactivation at 80 °C for 20 min.
The cleaned PCR product was sequenced with BigDye Terminators (Life Technologies) on an ABI3300xl according to standard protocols. The variant was screened in 326 unrelated normal Australian controls using the MassArray platform (Sequenom, California, USA) and iPlEX chemistry (Sequenom) at the Australian Genome Research Facility (QLD, Australia) and assessed for conservation across species using The Universal Protein Resource (UniProt) database (http://www.uniprot.org/).
Results
DNA from the proband was sequenced for 51 known congenital cataract genes using an Ion AmpliSeq custom amplicon panel. A total of 1,023,730 reads were mapped against the reference genome (hg19), of which 94.18% were on target. An average read depth of 841.9× was achieved for a total of 1216 amplicons with 96.05% of the target bases covered at least 20 fold. A total of 134 variants were annotated (Additional file 2), of which only six were novel or rare and nonsynonymous. Of the six variants, only one was not present in an in-house list of sequencing errors previously seen on this gene panel and predicted to be pathogenic by both SIFT and Polyphen-2 (five variants were false positive). This novel coding variant was a missense variant in the MAF gene (c.176C > G, p.(Pro59Arg)) (Fig. 1b). It was predicted to be pathogenic by SIFT and Polyphen-2 and is in a highly conserved region of the protein (Fig. 1c). The variant was also present in the affected mother and absent in 326 screened unrelated Caucasian controls. DNA was not available from the two unaffected siblings.
The 20-year-old proband (CS108.01) was diagnosed at birth with bilateral congenital cataract, described as nuclear and posterior polar in the right eye, and milder posterior polar oil droplet cataract in the left eye (Fig. 2a). Cataract in the right eye was removed at 5 months of age and the patient subsequently developed aphakic glaucoma. The right eye ultimately was significantly amblyopic. The proband also had mild to moderate sensorineural hearing loss (he did not appear to have a hearing impairment in early childhood). He was diagnosed with Asperger syndrome and borderline intellectual abilities in childhood. He attended a special school because of the combination of Asperger syndrome and visual/hearing impairment. In spite of this, he completed secondary education and went on to university, implying normal intellectual abilities. His childhood assessments of mild intellectual disability and borderline abilities are likely to have reflected the autism spectrum disorder and possibly the visual impairment. He developed scoliosis during teenage and had seizures at 13.5 years (two, 2 weeks apart). His height was 25th–50th percentile and head circumference was 50th–98th percentile. He had a distinctive facial appearance with narrow posteriorly rotated ears with upturned ear lobules, downslanting palpebral fissures, flat mid-face, short philtrum, prominent narrow chin and dental malocclusion (Fig. 2c). There was no joint limitation. The 53-year-old mother of the proband, CSA108.02, who had a mild learning disability and hearing impairment, had cataract extraction at the age of 40. She had not been diagnosed to have an autism spectrum disorder, had not had seizures and did not have joint limitation. Other features were normal height (10th–25th percentile), premature hair loss and double nails, with a nail growing out over the top of the existing one. She had mildly down slanting palpebral fissures, flat mid-face, a relatively prominent chin and widely spaced lower teeth (Fig. 2b).

Clinical features of the syndrome in family CSA108. a Phenotype of syndromic cataract in CSA108.01. Slit-lamp photographs showing posterior polar oil droplet cataract with posterior lenticonus. b Dental abnormalities in CSA108.01 (left) and CSA108.02 (right). c Facial features in CSA108.01 (left) and CSA108.02 (right). In particular, note flat mid-face in both, and short philtrum, long/narrow chin and upturned ear lobules in CSA108.01
Discussion
Undergoing traditional diagnostic assessment procedures, the clinical diagnosis of this proband with a rare syndromic congenital cataract phenotype was complicated and protracted. Past investigations included brain MRI, EEG, karyotyping, subtelomere FISH, FISH for Smith-Magenis syndrome, TORCH serology, urine metabolic screen for amino acids, organic acids and mucopolysaccharides, galactose-1-phosphate uridyl transferase, 7-dehydrocholesterol and very long chain fatty acids; all were normal apart from a diffusely abnormal EEG. Sequencing of the NHS gene associated with Nance-Horan syndrome (congenital cataract, dental anomalies and developmental delay) did not detect any pathogenic variants. The implementation of next generation sequencing including targeted gene sequencing panels such as PGM (Personal Genome Machine) results in more convenient molecular diagnostic process.
Although this study cannot entirely rule out a novel cause for disease, the features described in the proband and his mother are consistent with the condition previously reported independently by Aymé and Phillip [23] and Gripp et al. [24] (MIM 601088). There also have been reports of a similar syndrome by Fine and Lubinsky [25] and Preus et al. [26]. A recent study by Niceta et al. [27] reported a narrow spectrum of amino-acid substitutions within the MAF protein (Fig. 3), causing cataract, deafness, intellectual disability, seizures, a distinctive flat facial appearance, skeletal anomalies and reduced growth. The authors proposed the eponym Aymé-Gripp for this multisystem disorder. The reported de novo amino acid substitutions in MAF associated with this syndrome are p.(Ser54Leu), p.(Thr58Ala), p.(Thr58Ile), p.(Pro59His), p.(Pro59Leu), p.(Thr2Arg) and p.(Pro69Arg). Interestingly all these variants are located within the N-terminal transactivation domain of MAF as is the p.(Pro59Arg) substitution reported here (Fig. 3). Unlike other reported variants in MAF associated with Aymé-Gripp syndrome [27], the variant described here was inherited, with transmission from mother to son. Our findings also show that variants associated with Aymé-Gripp syndrome can display intra-familial variability since the mother had a substantially milder phenotype than the proband.

Schematic of the human MAF protein indicating the positions of reported variants (Adapted from Niceta et al. [27]). The protein contains an N-terminal transactivation domain and a C-terminal DNA binding domain. The C-terminal domain consists of an extended homology region, basic region (aa288–313) and leucine-zipper region (aa316–aa337). The variants associated with Aymé-Gripp syndrome are located in the N-terminal transactivation domain including the variant (p.(Pro59Arg)) reported here (bolded and underlined). Other variants are located within the C-terminal DNA-binding domain and are associated with other forms of congenital cataract mainly isolated
There also have been multiple other reports of variants in MAF associated with various forms of congenital cataract (Fig. 3): p.(Arg294Trp), p.(Lys297Arg), p.(Arg299Ser) and p.(Lys320Gly) variants have been linked with nuclear congenital cataract, [28] cerulean congenital cataract and microcornea [29], lamellar cataract with microcornea and iris coloboma, [30] and nuclear, punctate, stromal cataract with microcornea [3], respectively. Jamieson et al. described a family with juvenile onset progressive cataract of cortical pulverulent opacities with anterior and posterior sutural densities, anterior segment dysgenesis and microphthalmia associated with the cytogenetically balanced chromosome translocation 46, XY, t(5;16) (p15.3;q23.2), which transected the genomic-control domain of MAF [31]. They also reported a variant in the DNA-binding domain of MAF (p.(Arg288Pro)) in a three generations family with lamellar cortical and nuclear pulverulent cataract, microcornea, and iris coloboma. Narumi et al. [32] identified a MAF variant (p.(Gln303Leu)) through whole exome sequencing in a family with phenotypically variable congenital cataract (lamellar or anterior polar with microcornea and iris coloboma). The affected proband was diagnosed with lamellar cataract without any other eye malformation with language development delay and autism. The proband was also screened for variants in the NHS gene, however similar to the case we are reporting here, no variant in this gene was detected.
Many transcription factor genes are involved in lens development and their functions are important for proper lens induction, and cell proliferation and differentiation [33]. MAF belongs to the bZIP transcription factor family. It forms both homodimers and heterodimers, and binds to MAF response elements in target genes [7]. MAF is expressed in lens fibre cells during lens development and it has been demonstrated that homozygous Maf mutant mice had defective differentiation of lens fibre cells [34]. MAF has been proposed to regulate the expression of the lens specific genes including Crystallins [33, 35]. It has been demonstrated that the activity of the large MAF transcription factors is strongly dependent on phosphorylation within the conserved transactivation domain of the protein. This domain is rich in aspartic acid, glutamic acid, serine, threonine and proline residues [5, 6].
MAF is phosphorylated by glycogen synthase kinase 3 (GSK3), a serine/threonine protein kinase. Phosphorylation increases the transactivation activity of MAF and induces protein degradation [36]. GSK3 requires a priming phosphorylation on the substrate four amino acids C-terminal of the target phosphorylation sites [27, 37]. It has been demonstrated that the Thr58 residue is one of the GSK3 phosphorylation target sites [36] and residue Pro59 is located between the target (Thr58) and primed residues (Thr62) and is essential for GSK3 phosphorylation activity. Thus, substitution of this residue to Arginine, as seen in this family, might impair GSK3-mediated phosphorylation of MAF proteins, resulting in inefficient ubiquitination of the transcription factor and its decreased degradation and functional dysregulation. Consistently, this residue is highly conserved between species including mammals, birds, fish and amphibians, further suggesting its functional importance. Other variants at the same residue p.(Pro59His) and p.(Pro59Leu) [27] have been also reported to cause a similar phenotype.
Conclusion
We report a case of syndromic congenital cataract with similar features to those described by Niceta et al [27]. Ayme-Gripp syndrome’s key features are congenital cataracts, sensorineural hearing loss, intellectual disability, seizures, brachycephaly, flat face and short stature. The proband displayed all of the features except intellectual disability (though he has Asperger syndrome) and short stature. These features, combined with the novel identified variant in the transactivation domain of MAF, are consistent with a diagnosis of Aymé-Gripp syndrome in this family. This case is one of the most mildly affected reported and this is the first report of an inherited variant associated with this syndrome. This study shows the power and feasibility of next generation sequencing for variant detection in a clinically and genetically heterogeneous condition like syndromic congenital cataract, and demonstrates the ability of the technology to diagnose patients on the basis of their genetic results in combination with their phenotypic data.
Abbreviations
- ExAC:
-
Exome Aggregation Consortium
- MAF:
-
v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog
- PCR:
-
Polymerase Chain Reaction
- PGM:
-
Personal Genome Machine
References
Francis PJ, Berry V, Bhattacharya SS, Moore AT. The genetics of childhood cataract. J Med Genet. 2000;37:481–8.
Wirth MG, Russell-Eggitt IM, Craig JE, Elder JE, Mackey DA. Aetiology of congenital and paediatric cataract in an Australian population. Br J Ophthalmol. 2002;86:782–6.
Hansen L, Mikkelsen A, Nurnberg P, Nurnberg G, Anjum I, Eiberg H, Rosenberg T. Comprehensive mutational screening in a cohort of Danish families with hereditary congenital cataract. Invest Ophthalmol Vis Sci. 2009;50:3291–303.
Hejtmancik JF. Congenital cataracts and their molecular genetics. Semin Cell Dev Biol. 2008;19:134–49.
Tsuchiya M, Misaka R, Nitta K, Tsuchiya K. Transcriptional factors, Mafs and their biological roles. World J Diabetes. 2015;6:175–83.
Kataoka K. Multiple mechanisms and functions of maf transcription factors in the regulation of tissue-specific genes. J Biochem. 2007;141:775–81.
Churchill A, Graw J. Clinical and experimental advances in congenital and paediatric cataracts. Philos Trans R Soc Lond B Biol Sci. 2011;366:1234–49.
Aldahmesh MA, Khan AO, Mohamed JY, Hijazi H, Al-Owain M, Alswaid A, Alkuraya FS. Genomic analysis of pediatric cataract in Saudi Arabia reveals novel candidate disease genes. Genet Med. 2012;14:955–62.
Pras E, Raz J, Yahalom V, Frydman M, Garzozi HJ, Pras E, Hejtmancik JF. A nonsense mutation in the glucosaminyl (N-acetyl) transferase 2 gene (GCNT2): association with autosomal recessive congenital cataracts. Invest Ophthalmol Vis Sci. 2004;45:1940–5.
Azuma N, Hirakiyama A, Inoue T, Asaka A, Yamada M. Mutations of a human homologue of the Drosophila eyes absent gene (EYA1) detected in patients with congenital cataracts and ocular anterior segment anomalies. Hum Mol Genet. 2000;9:363–6.
Nonnenmacher L, Langer T, Blessing H, Gabriel H, Buchwald HJ, Meneksedag C, Kohne E, Gencik M, Debatin KM, Cario H. Hereditary hyperferritinemia cataract syndrome: clinical, genetic, and laboratory findings in 5 families. Klin Padiatr. 2011;223:346–51.
Shiels A, Bennett TM, Knopf HL, Yamada K, Yoshiura K, Niikawa N, Shim S, Hanson PI. CHMP4B, a novel gene for autosomal dominant cataracts linked to chromosome 20q. Am J Hum Genet. 2007;81:596–606.
Chen J, Ma Z, Jiao X, Fariss R, Kantorow WL, Kantorow M, Pras E, Frydman M, Pras E, Riazuddin S, et al. Mutations in FYCO1 cause autosomal-recessive congenital cataracts. Am J Hum Genet. 2011;88:827–38.
Jamieson RV, Farrar N, Stewart K, Perveen R, Mihelec M, Carette M, Grigg JR, McAvoy JW, Lovicu FJ, Tam PP, et al. Characterization of a familial t(16;22) balanced translocation associated with congenital cataract leads to identification of a novel gene, TMEM114, expressed in the lens and disrupted by the translocation. Hum Mutat. 2007;28:968–77.
Lachke SA, Alkuraya FS, Kneeland SC, Ohn T, Aboukhalil A, Howell GR, Saadi I, Cavallesco R, Yue Y, Tsai AC, et al. Mutations in the RNA granule component TDRD7 cause cataract and glaucoma. Science. 2011;331:1571–6.
Aldahmesh MA, Khan AO, Mohamed JY, Alghamdi MH, Alkuraya FS. Identification of a truncation mutation of acylglycerol kinase (AGK) gene in a novel autosomal recessive cataract locus. Hum Mutat. 2012;33:960–2.
Zhou G, Zhou N, Hu S, Zhao L, Zhang C, Qi Y. A missense mutation in CRYBA4 associated with congenital cataract and microcornea. Mol Vis. 2010;16:1019–24.
Ferda Percin E, Ploder LA, Yu JJ, Arici K, Horsford DJ, Rutherford A, Bapat B, Cox DW, Duncan AM, Kalnins VI, et al. Human microphthalmia associated with mutations in the retinal homeobox gene CHX10. Nat Genet. 2000;25:397–401.
Reis LM, Tyler RC, Volkmann Kloss BA, Schilter KF, Levin AV, Lowry RB, Zwijnenburg PJ, Stroh E, Broeckel U, Murray JC, et al. PITX2 and FOXC1 spectrum of mutations in ocular syndromes. Eur J Hum Genet. 2012;20:1224–33.
Hughes AE, Bradley DT, Campbell M, Lechner J, Dash DP, Simpson DA, Willoughby CE. Mutation altering the miR-184 seed region causes familial keratoconus with cataract. Am J Hum Genet. 2011;89:628–33.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Ayme S, Philip N. Fine-Lubinsky syndrome: a fourth patient with brachycephaly, deafness, cataract, microstomia and mental retardation. Clin Dysmorphol. 1996;5:55–60.
Gripp KW, Nicholson L, Scott Jr CI. Apparently new syndrome of congenital cataracts, sensorineural deafness, Down syndrome-like facial appearance, short stature, and mental retardation. Am J Med Genet. 1996;61:382–6.
Fine BA, Lubinsky M. Craniofacial and CNS anomalies with body asymmetry, severe retardation, and other malformations. J Clin Dysmorphol. 1983;1:6–9.
Preus M, Cooper AR, O’Leary E. Sensorineural hearing loss, small facial features, submucous cleft palate, and myoclonic seizures. J Clin Dysmorphol. 1984;2:30–1.
Niceta M, Stellacci E, Gripp KW, Zampino G, Kousi M, Anselmi M, Traversa A, Ciolfi A, Stabley D, Bruselles A, et al. Mutations Impairing GSK3-Mediated MAF Phosphorylation Cause Cataract, Deafness, Intellectual Disability, Seizures, and a Down Syndrome-like Facies. Am J Hum Genet. 2015;96:816–25.
Sun W, Xiao X, Li S, Guo X, Zhang Q. Exome sequencing of 18 Chinese families with congenital cataracts: a new sight of the NHS gene. PLoS One. 2014;9:e100455.
Vanita V, Singh D, Robinson PN, Sperling K, Singh JR. A novel mutation in the DNA-binding domain of MAF at 16q23.1 associated with autosomal dominant “cerulean cataract” in an Indian family. Am J Med Genet A. 2006;140:558–66.
Hansen L, Eiberg H, Rosenberg T. Novel MAF mutation in a family with congenital cataract-microcornea syndrome. Mol Vis. 2007;13:2019–22.
Jamieson RV, Perveen R, Kerr B, Carette M, Yardley J, Heon E, Wirth MG, van Heyningen V, Donnai D, Munier F, et al. Domain disruption and mutation of the bZIP transcription factor, MAF, associated with cataract, ocular anterior segment dysgenesis and coloboma. Hum Mol Genet. 2002;11:33–42.
Narumi Y, Nishina S, Tokimitsu M, Aoki Y, Kosaki R, Wakui K, Azuma N, Murata T, Takada F, Fukushima Y, et al. Identification of a novel missense mutation of MAF in a Japanese family with congenital cataract by whole exome sequencing: a clinical report and review of literature. Am J Med Genet A. 2014;164A:1272–6.
Huang B, He W. Molecular characteristics of inherited congenital cataracts. Eur J Med Genet. 2010;53:347–57.
Kawauchi S, Takahashi S, Nakajima O, Ogino H, Morita M, Nishizawa M, Yasuda K, Yamamoto M. Regulation of lens fiber cell differentiation by transcription factor c-Maf. J Biol Chem. 1999;274:19254–60.
Ring BZ, Cordes SP, Overbeek PA, Barsh GS. Regulation of mouse lens fiber cell development and differentiation by the Maf gene. Development. 2000;127:307–17.
Herath NI, Rocques N, Garancher A, Eychene A, Pouponnot C. GSK3-mediated MAF phosphorylation in multiple myeloma as a potential therapeutic target. Blood Cancer J. 2014;4:e175.
Dajani R, Fraser E, Roe SM, Young N, Good V, Dale TC, Pearl LH. Crystal structure of glycogen synthase kinase 3 beta: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell. 2001;105:721–32.
Acknowledgements
We all express our appreciations to the family members who kindly gave consent for this finding to be presented in this paper.
Funding
This work was supported by funding from the Channel 7 Children’s Research Foundation, the Ophthalmic Research Institute of Australia, and a Centre for Research Excellence Grant from the National Health and Medical Research Council (NHMRC) of Australia. JEC is supported by an NHMRC Practitioner Fellowship and KPB by an NHMRC Senior Research Fellowship.
Availability of data and materials
A list of all variants annotated in the screened proband is available in Additional file 2. All other materials supporting the finding of this study are provided within the main article (in figures) and in Additional file 1: Table S1.
Authors’ contributions
SJ, JEC, SS, ES, KML, EH, and KPB contributed to the conception, design and interpretation of data. SJ carried out the molecular genetic studies, data analysis, and was in charge of manuscript preparation. KPB has been involved in drafting the manuscript critically. JEC, SS, ES, KML and EH also helped with the manuscript preparation. JEC, TC and EH helped with patient recruitment and clinical information. All authors have read and approved the final version of the manuscript. None of the authors have any competing interests.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Written informed consent was obtained from participants for publication of this result and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal. Where participants were minor (under the age of 18) or unable to personally provide consent, written informed consent was obtained from the parent or legal guardian.
Ethics approval and consent to participate
The study adhered to the tenets of the Declaration of Helsinki and was approved by the Southern Adelaide Clinical Human Research Ethics Committee. Written informed consent was obtained from all of the family members and/or their guardians for participation in this research.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
List of reported paediatric cataract genes selected for sequencing in this study. (DOCX 39 kb)
Additional file 2:
List of annotated and selected variants. (XLSX 41 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Javadiyan, S., Craig, J.E., Sharma, S. et al. Novel missense mutation in the bZIP transcription factor, MAF, associated with congenital cataract, developmental delay, seizures and hearing loss (Aymé-Gripp syndrome). BMC Med Genet 18, 52 (2017). https://doi.org/10.1186/s12881-017-0414-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-017-0414-7