
Abstract
Background
Hemophagocytic lymphohistocytosis (HLH) is a rare but fatal hyperinflammatory syndrome caused by uncontrolled proliferation of activated macrophages and T lymphocytes secreting high amounts of inflammatory cytokines. Genetic defect is a common cause of HLH. HLH is complicated to be diagnosed as there are many common symptoms with other disorders.
Case presentation
Here we report on an HLH case caused by 1 bp deletion in gene SH2D1A. Patient was a 3-years-old boy and had fever for more than 8 days. Splenomegaly and hemophagocytosis in bone marrow were observed in examination. The results of the blood analysis suggested the diagnosis of HLH. Genetic test based on high throughput amplicon sequencing was then conducted by targeting all six known HLH-causing genes simultaneously. It took only one single day to accomplish the amplicon sequencing library preparation, sequencing and data analysis. Finally, a novel 1 bp deletion in gene SH2D1A was discovered. The result was also confirmed by Sanger sequencing. The result of the genetic test served as a good basis for further diagnosis of HLH.
Conclusion
This is the first case that the disease-causing genetic defect of HLH was quickly determined by high throughput amplicon sequencing. This diagnosis was also confirmed by Sanger sequencing and cross-validated by blood analysis and other clinical criteria. This case suggests that genetic test based on amplicon sequencing is a powerful tool for diagnosis of HLH and other diseases caused by genetic defect.
Similar content being viewed by others

Background
Hemophagocytic lymphohistocytosis (HLH) is a potentially fatal disease with severe systemic syndrome, which is caused by uncontrolled proliferation of activated macrophages and T lymphocytes, and impaired regulation of the immune system. The first case was reported at 1952 by Farquhar et al., and the mortality rate of HLH ranged from 22 to 60% [1, 2]. Many reports have revealed that HLH could affect all age groups, from preterm neonates to elderly adults [3]. The overall occurrence rate might be underestimated as it could be diagnosed as other disease sharing similar symptom [3, 4].
Usually HLH is rapidly fatal unless treated aggressively [5]. Rapid definitive diagnosis and appropriate treatment are extremely important for life-saving and improved prognosis for patients. Established clinical and laboratory criteria help significantly in accurate clinical diagnosis, but most of them are time-consuming. Furthermore, they are not sensitive and specific enough for HLH diagnosis, so the results of these tests may be supportive of, but not diagnostic of, HLH. Therefore, a gene mutation analysis should be the gold standard for making a definitive diagnosis. To date, a total of 6 causative genes have been identified, including SH2D1A, PRF1, UNC13D, STX11, STXBP2 and XIAP, and a few mutations in these genes have been reported to be the common cause of HLH [6–9]. Usually, these genes were checked one by one to detected mutations. Here, high throughput genetic analysis was conducted to detect mutations in all six genes simultaneously with amplicon sequencing in a HLH patient. We identified a novel mutation in gene SH2D1A, which was also confirmed by Sanger sequencing and this patient was diagnosed as HLH finally. This indicated that amplicon sequencing was a powerful tool in diagnosis of genetic disease, especially for cases involved multiple candidate causative genes.
Case presentation
The study was approved by the institute ethics committee of Key Laboratory of Birthdefects Prevention, National Health and Family Planning Commission, Zhengzhou, China. Informed consent from the parents was obtained before collecting blood samples. A 3-years-old boy with HLH was diagnosed based on blood analysis and genetic detection. The causing mutation was a homozygous 1 bp deletion (c.92delT) in the first exon of SH2D1A and this deletion was validated by Sanger sequencing. This patient and his parents are all Han Chinese from Henan province of China.
Clinical data
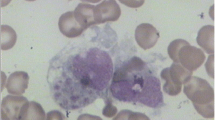
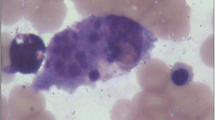
The 3-years-old patient had fever for more than 8 days with unknown origin when he was admitted to Key Laboratory of Birthdefects Prevention, National Health and Family Planning Commission, Zhengzhou, China. He had taken penicillin for 5 days before admission. On examination, the temperature was measured to 39.5 °C. No other respiratory symptoms but tachypnea presented in physical examination. The central nervous system was normal. Ultrasound examination revealed splenomegaly. Blood analysis was conducted at Key Laboratory of Birthdefects Prevention and hemoglobin was measured to be 90 g/L. Platelets, Neutrophils and Fibrinogen were measured to be 22× 109/L, 7.54× 109/L and 3.42 g/L, respectively. Low NK-cell activity (5.05%) and low Plasma albumin (18.9 g/L) were also observed. The bone marrow examination suggested hemophagocytosis without any evidence of malignancy [Fig. 1]. The results of the blood analysis and clinical features suggested the diagnosis of HLH based on HLH-2004 guidelines [10]. A two-generation pedigree including the patient and his parents was generated.

The bone marrow examination. Phagocytosis could be clearly observed in the boon marrow
Detection of mutation of six genes
Genomic DNA was purified from peripheral blood mononuclear cells PBMC with QIAamp Blood Kit (Qiagen, Hilden, Germany) according to the protocol. Multiplexed primers were designed for 6 disease-causing genes (SH2D1A, PRF1, UNC13D, STX11, STXBP2 and XIAP) with Ion AmpliSeq™ Designer (https://www.ampliseq.com) and a total of 64 exons were covered by these primers, including the whole coding regions of these genes and splicing sites for each exon-intron boundaries. Multiplexed polymerase chain reaction (PCR) amplification was conducted and the PCR product was ligated with adapters, purified and then sequenced by proton sequencing based S5XL genetic analyzer (Applied Biosystems®, Life Technologies, Grand Island, NY, USA) and mutations were analyzed using VariantCaller V1.0 software embedded in the Torrent Suited software (Life Technologies). Primer information could be found in supplementary file [see Additional file 1].
The average base coverage depth for the whole target regions was 1,515 and more than 97.13% of all sites had coverage of more than 100-fold, which indicated a good performance of multiplexed PCR. A total of 19 SNPs (17 heterozygous SNPs and two homozygous SNPs) were detected in all five genes except XIAP and no insertion was found in all genes [Table 1]. One homozygous deletion was identified in the first exon of SH2D1A. Among all 19 SNPs, 11 were known mutations recorded in the NCBI refSNP database and no known cases have been reported to be associated with these SNPs. For the remaining eight novel SNPs, no damage-causing effect was found by searching literatures and databases, as well as domain analysis for each protein, including six synonymous mutations and two missense mutations (c.2599A > G in the 27th exon of gene UNC13D, c.1576A > G in the 18th exon of gene STXBP2). In summary, no novel and known SNPs had been proved to be disease-related mutations.
A novel homozygous 1 bp deletion (c.92delT) was identified in exon 1 of SH2D1A, which was located on the X chromosome (at Xq25) [11]. This deletion was confirmed by Sanger sequencing and also detected in the patient’s mother, but not in his father [Fig. 2a]. This indicated that it was not a de novo variation, but inherited from the mother. Interestingly, this variation in the mother was found to be heterozygous based on Sanger sequencing [Fig. 2a]. The deletion resulted in an introduction of a premature stop codon and produced a truncated protein (79aa, p.Leu31Argfs*50) [Fig. 2b]. SH2D1A is a signaling lymphocytic activation molecule (SLAM)-associated protein (SAP), and it is one of the known disease-causing genes associated with immunodeficiency, which led to X-linked lymphoproliferative disease (XLP) [12]. The affect of the mutations occurred in SH2D1A typically manifested in childhood or early adolescence and it was estimated that it affected 1–3 per million boys [13]. A lot of mutations in SH2D1A have been identified and more than 200 cases related with these mutations has been reported [14, 15]. We found three mutational hotspots in the SH2D1A gene according to the records in SWISSPROT. The novel 1 bp deletion (c.92delT) presented here was located in the first one of these mutational hotspots.

Mutations in the gene SH2D1A. a Sanger sequencing results of the 3-years-old child (underside), his father (top left) and his mother (top right). Mutation position is marked with red arrow. The results show heterozygosity for this mutation in his mother, but wild type sequences in his father. As indicated, deleted nucleotide caused a frameshift in the corresponding sequence. b) Mutations recorded in SWISSPROT. All mutations are marked in blue and each mutational hotspot is indicated by black box. Almost all the mutations located in these three regions have been reported to be disease-causing defects. The novel deletion detected in this study is marked with red arrow and the premature stop codon is marked with purple arrow. “VAR_048***” is the accession No in SWISSPROT, “ND” represents for “Not detected”, “+” indicates that the mutation could reduce protein stability or interaction with other protein, “++” indicates a significant reduction and “N” indicates totally abolished interaction with SLAMF1. “-“means no data available
Discussion
In this case, two other possible diseases have been noted considering that SH2D1A mutation could involve in rheumatoid arthritis (RA) [16] and systemic lupus erythematosus [17], but both of these diseases were excluded. Firstly, splenomegaly and Hemophagocytosis in bone marrow were observed in examination, and all blood analysis showed that some key indexes fitted in with the diagnosis of HLH based on HLH-2004 guidelines, which has been revised at 2007. If the child had RA, the temperature will not be as high as 39.5 °C, but just low-grade fever. And it should not be systemic lupus erythematosus, because no obvious symptoms could be found in physical examination. The central nervous system was also normal. Together with the identification of 1 bp deletion in exon of SH2D1A, a diagnosis of HLH could be accepted. However, it is interesting to note that the gene SH2D1A is located on the X chromosome [11] and Sanger sequencing revealed that the mutation was heterozygous in the mother, which might explain why his mother had no obvious symptoms of HLH. This also indicated that the case presented here was X-linked genetic disease, especially considering the incidence of the patient. As soon as sequencing result was acquired and HLH diagnosis was confirmed, the patient received full course of dexamethasone combining with VP-16 chemotherapy, according to the guideline of HLH-2004 protocol [10]. The clinical manifestation quickly stabilized and currently he is under treatment for further HSCT procedure.
SH2D1A encodes the signaling lymphocytic activation molecule (SLAM)-associated protein (SAP), which interacts with members of the CD2 subset of the Ig superfamily of cell-surface receptors [12]. It has been reported that there are three putative acting mechanisms that could cause XLP due to mutation in SH2D1A, including reduced gross expression, reduced ability binding to SLAM family receptors and inability to activate signal transduction downstream of the SLAM receptor–SAP complex efficiently [18]. In the present case, it is believed that no functional protein was generated due to deletion of 92th base (T), which could lead to functional defects in natural killer (NK) cells. NK cells are required for clearance of viral infection as well as regulation and termination of the inflammatory response [19]. Phagocytes, neutrophils and NK cells constitute the innate immune system and persistent antigen exposure will keep them active. These activated cells released proimflammatory cytokines such as TNF-α, IL-1 and IL-6, which could cause fever, hyperferritinemia and other symptoms.
HLH occurs predominantly in infants and very young children. Based on the mechanism of morbidity, HLH can either be genetic, or acquired due to viral infections, malignancies or rheumatic diseases. According to classification of histiocytic disorders, HLH is subdivided into familial HLH (FHLH) and secondary HLH (sHLH) [2]. Based on genetic etiology, FHLH has been subcategorized into 5 subtypes, i.e., FHLH-1 to FHLH-5 [20]. However, these disorders may be difficult to be distinguished from each other clinically. For example, the clinical symptoms of familial HLH usually become evident within the first 2 months of life, but most clinicians cannot distinguish FHL from other disorders if the patient is the first child or in the absence of affected siblings.
A set of clinical and laboratory criteria have been established for accurate diagnose of HLH [10], such as prolonged high-grade fever, progressive cytopenias, hepatosplenomegaly with liver dysfunction, skin rash, coagulopathy and variable neurologic symptoms, and these criteria have been widely used in clinical practice. The pathologic findings of activated macrophages, engulfing erythrocytes, leukocytes, platelets and their precursor cells were also important for diagnosis of HLH. However, not all HLH cases present these clinical manifestations in the initial stage [21], and the organs involved may be different [22]. Especially, it is has been reported that HLH may also commonly occurred in a setting of rheumatologic illness [23].
In practice, analysis of bone marrow aspirate is commonly used for HLH diagnosis, which only has a sensitivity of 60% [24]. Therefore, a negative bone marrow analysis result should not completely rule out HLH. A set of other rapid diagnostic tests have been proposed to aid the diagnosis of HLH, including Ferritin, soluble IL-2 receptor, IFN-g, IL-10 and other proinflammatory cytokines [10, 25, 26]. All these indices have been reported to be useful in diagnosing HLH. Novel methods have also been developed for the early and rapid detection of patients. For example, flow cytometry assay has been used to evaluate the expression of lymphoid SAP (XLP1), perforin and XIAP (XLP2) [27], and it could also detect revertants and somatic mutations [28]. It has to be noted that all these criteria and methods are proposed based on physiological indices, not genetic, and they have limited sensitivity and specificity for HLH. On the other side, screening of mutations in all these 6 causative genes are generally conducted by Sanger sequencing, and constrained by its low throughput. Now high throughput sequencing is cost-effective and time-effective for genetic testing, and it could detect all target loci simultaneously in 1 day, even most proportion of whole genome if necessary. Together with highly multiplexed PCR technology, it could identify all kinds of variants with high accurate by covered the target region (i.e., causative genes) with a proper depth. This may help to reduce the time consumed by differential diagnosis and make the definitive diagnosis rapidly. This is extremely important for life-saving and getting improved prognosis by giving patients appropriate treatment as soon as possible. In order to prevent misdiagnosis and give as much information as possible for clinicians, it will be helpful and necessary to do this genetic analysis immediately with amplicon sequencing after child is born.
Conclusion
In summary, 6 HLH-related genes were tested at once in a suspected HLH patient and a novel homozygous 1 bp deletion (c.92delT) in exon 1 of SH2D1A was indentified. The diagnosis of HLH was further confirmed by this result. Most HLH occurred in childhood and the survival rate is very poor, even with treatment [29]. Rapid definitive diagnosis and appropriate treatment are extremely important for life-saving and prognosis-improving. This indicated that amplicon sequencing is necessary for rapid and accurate screening of mutations in the diagnosis of genetic disease, especially those clinically indistinguishable disorders. Other methods based on DNA and proteins analysis have been reported for the detection of SH2D1A mutation [28, 30], but none could be used to detect all possible genes simultaneously and rapidly. Ultimately, this case proposed a novel method to identify HLH-related mutations, and improve the understanding of their roles during the regulation of host antiviral immune responses. It will also facilitate development of novel therapies for these rare but devastating disorders.
Abbreviations
- FHLH:
-
Familial hemophagocytic lymphohistocytosis
- HLH:
-
Hemophagocytic lymphohistocytosis
- sHLH:
-
Secondary hemophagocytic lymphohistocytosis
- XLP:
-
X-linked lymphoproliferative disease
References
Farquhar JW, Claireaux AE. Familial haemophagocytic reticulosis. Arch Dis Child. 1952;27(136):519–25.
Lin YH, Lin YH, Shi ZY. A case report of scrub typhus-associated hemophagocytic syndrome and a review of literature. Jpn J Infect Dis. 2014;67(2):115–7.
Suzuki N, Morimoto A, Ohga S, Kudo K, Ishida Y, Ishii E. Characteristics of hemophagocytic lymphohistiocytosis in neonates: a nationwide survey in Japan. J Pediatr. 2009;155:235–8.
Tabata R, Tabata C, Terada M, Nagai T. Hemophagocytic syndrome in elderly patients with underlying autoimmune diseases. Clin Rheumatol. 2009;28:461–4.
Erdős M, Uzvölgyi E, Nemes Z, et al. Characterization of a disease-causing mutation of SH2D1A in a family with X-linked lymphoproliferative disease. Hum Mutat. 2005;25:506.
Rigaud S, Fondane’che MC, Lambert N, Pasquier B, Mateo V, Soulas P, Galicier L, Le Deist F, Rieux-Laucat F, Revy P, Fischer A, de Saint Basile G, Latour S. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. 2006;444:110–4.
Meeths M, Chiang SCC, Wood SM, Entesarian M, Schlums H, Bang B, et al. Familial hemophagocytic lymphohistiocytosis type 3 (FHL3) caused by deep intronic mutation and inversion in UNC13D. Blood. 2011;118:5783–93.
Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood. 2011;118:5794–8.
Liu J, Tian W, Wang F, Teng W, Zhang Y, Tong C, Zhang C, Ju Y, Zhang B, Zhao S, Liu H. Maternal onset de novo SH2D1A mutation and lymphocytic choriomeningitis virus infection in a patient with X-linked lymphoproliferative disease type 1: a case report. Mol Med Rep. 2015;11(5):3291–4.
Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–31.
Sayos J, Wu C, Morra M, et al. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature. 1998;395(6701):462–9.
Nichols KE, Harkin DP, Levitz S, Krainer M, Kolquist KA, Genovese C, et al. Inactivating mutations in an SH2 domain-encoding gene in X-linked lymphoproliferative syndrome. Proc Natl Acad Sci U S A. 1998;95(23):13765–70.
Sumegi J, Huang DL, Lanyi A, Davis JD, Seemayer TA, Maeda A, Klein G, Seri M, et al. Correlation of mutations of the SH2D1A gene and Epstein-Barr virus infection with clinical phenotype and outcome in X-linked lymphoproliferative disease. Blood. 2000;96(9):3118–25.
Halasa NB, Whitlock JA, McCurley TL, et al. Fatal hemophagocytic lymphohistiocytosis associated with Epstein-Barr virus infection in a patient with a novel mutation in the signaling lymphocytic activation molecule-associated protein. Clin Infect Dis. 2003;37:e136.
Nichols KE, Ma CS, Cannons J, Schwartzberg PL, et al. Molecular and cellular pathogenesis of X-linked lymphoproliferative disease. Immunol Rev. 2005;203:180–99.
Takei M, Ishiwata T, Mitamura K, Fujiwara S, Sasaki K, Nishi T, Kuga T, Ookubo T, Horie T, Ryu J, Ohi H, Sawada S. Decreased expression of signaling lymphocytic-activation molecule-associated protein (SAP) transcripts in T cells from patients with rheumatoid arthritis. Int Immunol. 2001;13(4):559–65.
Furukawa H, Kawasaki A, Oka S, Shimada K, Matsui T, Ikenaka T, Hashimoto A, et al. Association of a single nucleotide polymorphism in the SH2D1A intronic region with systemic lupus erythematosus. Lupus. 2013;22(5):497–503.
Hare NJ, Ma CS, Alvaro F, et al. Missense mutations in SH2D1A identified in patients with X-linked lymphoproliferative disease differentially affect the expression and function of SAP. Int Immunol. 2006;18:1055–65.
Lykens JE, Terrell CE, Zoller EE, Risma K, Jordan MB. Perforin is a critical physiologic regulator of T-cell activation. Blood. 2011;118:618–26.
Janka GE, Schneider EM. Modern management of children with haemophagocytic lymphohistiocytosis. Br J Haematol. 2004;124:4–14.
Gholam C, Grigoriadou S, Gilmour KC, Gaspar HB. Familial haemophagocytic lymphohistiocytosis: advances in the genetic basis, diagnosis and management. Clin Exp Immunol. 2011;163:271–83.
Danhaive O, Caniglia M, Devito R, Piersigilli F, Corchia C, Auriti C. Neonatal liver failure and haemophagocytic lymphohistiocytosis caused by a new perforin mutation. Acta Paediatr. 2010;99:778–80.
Stephan JL, Zeller J, Hubert P, Herbelin C, Dayer JM, Prieur AM. Macrophage activation syndrome and rheumatic disease in childhood: a report of four new cases. Clin Exp Rheumatol. 1993;11(4):451–6.
Gupta A, Weitzman S, Abdelhaleem M. The role of hemophagocytosis in bone marrow aspirates in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;50:192–4.
Bleesing J, Prada A, Siegel DM, Villanueva J, Olson J, Ilowite NT, et al. The diagnostic significance of soluble CD163 and soluble interleukin-2 receptor alpha-chain in macrophage activation syndrome and untreated new-onset systemic juvenile idiopathic arthritis. Arthritis Rheum. 2007;56:965–71.
Xu XJ, Tang YM, Song H, Yang SL, Xu WQ, Zhao N, et al. Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children. J Pediatr. 2012;160:984–90.
Marsh RA, Bleesing JJ, Filipovich AH. Using flow cytometry to screen patients for Xlinked lymphoproliferative disease due to SAP deficiency and XIAP deficiency. J Immunol Methods. 2010;362:1–9.
Zhao M, Kanegane H, Kobayashi C, Nakazawa Y, Ishii E, Kasai M, et al. Early and rapid detection of X-linked lymphoproliferative syndrome with SH2D1A mutations by flow cytometry. Cytometry B Clin Cytom. 2011;80(1):8–13.
Seemayer TA, Gross TG, Egeler RM, Pirruccello SJ, Davis JR, Kelly CM, Okano M, Lanyi A, Sumegi J. X-linked lymphoproliferative disease: Twenty-five years after the discovery. Pediatr Res. 1995;38:471–8.
Gilmour KC, Cranston T, Jones A, et al. Diagnosis of X-linked lymphoproliferative disease by the analysis of SAP expression. Eur J Immunol. 2000;30:1691–7.
Acknowledgement
The authors also gratefully acknowledge the patients and their family for their participation in this research study. This work was supported by the Taihe Hospital and National Health and Family Planning Commission. We are grateful to Zhiwei Fang for suggestion about the analysis and meaningful discussion.
Funding
Not applicable.
Availability of data and materials
The sequence data have been deposited into NCBI Short Read Archive under accession number SRR4419660.
Authors’ contributions
MHY, GMF and QZ performed the experiments, GB and ZY finished the blood analysis and analyzed the data, ZSY analysis data and wrote the manuscript. ZSY, MHY and QGF design this study and edited the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Ethics approval and consent to participate
This study was approved according to the guidelines of the Committee on the Use of Human Subjects in Key Laboratory of Birthdefects Prevention. Informed consent from the parents was obtained before collecting blood samples.
Author information
Authors and Affiliations
Corresponding authors
Additional file
Additional file 1
Table S1: List of primers that were used for amplicon sequencing. (XLSX 19 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhou, S., Ma, H., Gao, B. et al. Characterization of a novel disease-causing mutation in exon 1 of SH2D1A gene through amplicon sequencing: a case report on HLH. BMC Med Genet 18, 15 (2017). https://doi.org/10.1186/s12881-017-0376-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-017-0376-9