
Abstract
Background
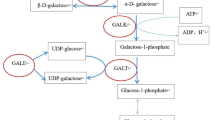
Classic galactosemia (OMIM #230400) is an autosomal recessive metabolic disorder caused by a deficiency of the galactose-1-phosphate uridyltransferase (GALT, EC2.7.7.12) protein due to mutations in the GALT gene. The aim of this study was to provide a comprehensive and updated mutation spectrum of GALT in a Korean population.
Methods
Thirteen unrelated patients screened positive for galactosemia in a newborn screening program were included in this study. They showed a reduced GALT enzyme activity in red blood cells. Direct sequencing of the GALT gene and in silico analyses were done to evaluate the impact of novel variations upon GALT enzyme activity. We also reviewed previous reports for GALT mutations in Koreans.
Results
We identified six novel likely pathogenic variations including three missense (p.Ala101Asp, p.Tyr165His, and p.Pro257Thr), one small deletion/insertion [c.826_827delinsAA (p.Ala276Asn)], one frameshift (p.Asn96Serfs*5), and one splicing (c.378-1G > C) likely pathogenic variations. The most frequent variation was the Duarte variant (c.940A > G, 35.3%), followed by c.507G > C (p.Gln169His, 9.6%), among 34 Korean patients. Other mutations were widely scattered. None of the eight common mutations used for targeted mutation analysis in Western countries including p.Gln188Arg, p.Ser135Leu, p.Lys285Asn, p.Leu195Pro, p.Tyr209Cys, p.Phe171Ser, c.253-2A > G, and a 5 kb deletion, had been found in Koreans until this study.
Conclusions
Considering the mutation spectrum in Koreans, direct sequence analysis of entire GALT exons is recommended for accurate diagnosis. The mutations responsible for GALT deficiency in the Korean population were clearly different from those of other populations.
Similar content being viewed by others


Background
Classical galactosemia (OMIM #230400) is an autosomal recessive inborn error of metabolism caused by a deficiency of the galactose-1-phosphate uridyltransferase (GALT, EC2.7.7.12) protein, which is encoded by the GALT gene. GALT maps to chromosome 9p13 and includes 11 exons that span about 4 kb of genomic DNA [1]. Galactosemia is a potentially fatal disease that manifests within the first week of life with poor feeding, jaundice, vomiting, liver dysfunction, increased bleeding tendency, and septicemia, leading to death if untreated [2]. It is well known that early detection of galactosemia and management with dietary restriction of galactose can prevent infants from a deteriorating state and leads to rapid clinical improvement. Many patients develop long-term complications, with a broad range of clinical symptoms such as cataracts, speech defects, poor growth, poor intellectual function, neurologic deficits, and premature ovarian insufficiency [3]. Many countries have implemented newborn screening programs in order to detect and manage the disease earlier [4]. A wide range of galactosemia frequency was estimated in Western countries (1:14,000 to 1:80,000) [4],[5] and much lower frequency reported in Asian populations [6],[7]. To date, more than 230 mutations responsible for galactosemia have been identified and updated in the GALT gene databases [8],[9]. Eight common GALT mutations for targeted mutation analysis have been introduced in Western countries, which include p.Gln188Arg, p.Ser135Leu, p.Lys285Asn, p.Leu195Pro, p.Tyr209Cys, p.Phe171Ser, c.253-2A > G, and a 5 kb deletion [10]-[12]. However, none of these mutations have been reported in Asian populations. Since there is a wide range of mutations among different ethnic background, genetic analysis is needed to understand the mutation spectrum of GALT in the Korean population. In Korea, a nationwide newborn screening program for galactosemia, first introduced in 1991, is now performed on more than 90% of Korean newborns [13]. Only 21 Korean patients with GALT-deficient galactosemia have been reported, which suggests a low prevalence [14],[15]. The aim of this study was to characterize and update the spectrum of GALT gene variations in Korean patients using direct sequence analysis and to produce a comprehensive literature review of GALT mutations in the Korean population.
Methods
Patients
Thirteen unrelated Korean children with abnormal newborn screening results for galactosemia and reduced GALT activities between April 2010 and April 2013 from two institutions (Samsung Medical Center and Soonchunhyang University Hospital) were included in this study. Ninety-six healthy subjects were also included as controls in order to exclude common polymorphisms for six novel variations. Additionally, a comprehensive review of the literature with previously reported GALT mutations in Koreans was conducted. This research was approved by the Samsung Medical Center Institutional Review Board and written informed consent was obtained from all subjects and/or their parents whenever possible.
GALT enzyme activity in red blood cells
GALT enzyme activity in red blood cells was measured by an established radiometric assay [16] and was expressed in μmol/hour (hr)/g hemoglobin (Hb). The reference range of GALT enzyme activity is from 20 to 35 μmol/hr/g Hb.
DNA amplification and direct sequence analysis
Human genomic DNA was prepared from frozen white blood cells using a Wizard genomic DNA purification kit (Promega, Madison, WI, USA) according to the manufacturer’s recommendations. All 11 exons and the flanking regions of the GALT gene were amplified by polymerase chain reaction (PCR) using primers designed by the authors (sequences available upon request) and a thermal cycler (Model 970; Applied Biosystems, Foster City, CA, USA). Direct sequencing of the DNA was performed using the ABI Prism 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) with the BigDye Terminator Cycle Sequencing-Ready Reaction Kit (Applied Biosystems Foster City, CA, USA). Nucleotides are numbered from the first adenine of the ATG translation initiation codon in the GALT cDNA Reference Sequence NM_000155.2.
Pathologic predictions of novel substitution variants
We performed in silico analyses to identify the mutational status of a novel exonic variation using two different servers. PolyPhen-2 prediction was used to identify the functional effect of human non-synonymous single nucleotide polymorphisms (http://genetics.bwh.harvard.edu/pph2/) and the second prediction program was SIFT (Sorting Intolerant From Tolerant, http://sift.jcvi.org/). “Damaging change” in PolyPhen-2 and “not tolerated” in SIFT both mean that the missense variation may be a mutation rather than a polymorphism. We also used the Human Splicing Finder (http://www.umd.be/HSF/) to predict any difference in splicing between the novel mutants and their respective wild-type reference sequences to estimate the mutation status of novel substitution variants. If in silico analyses predicted the novel variants to be likely pathogenic, then the novel variations are defined as likely pathogenic variations rather than detrimental mutations because expression studies were not performed.
Genetic analysis of normal individuals
The tests targeted the six novel sequence variations revealed in this study and were conducted on 192 alleles from 96 healthy adult individuals without any clinical symptoms of galactosemia. All genetic analyses were done using direct sequence analysis, as explained above.
Statistical tests
We applied the Kruskal-Wallis test with post-hoc analysis for comparison of patients with different variant alleles for the GALT gene including the Duarte variant using the MedCalc program for Windows, version 12.2.1 (MedCalc Software, Mariakerke, Belgium). A p value of less than 0.05 was considered significant.
Results
Demographics of GALT-deficient galactosemia
During the 3-year study period, 13 unrelated patients with GALT deficiency (six males and seven females) were included in this study. The median age at molecular diagnosis in the 13 Korean galactosemia subjects was 2.9 months (range, 1 month – 23 months). All but one had no significant clinical symptoms or signs of galactosemia. Only one patient had jaundice and cataracts whose age at molecular diagnosis was 23 months. All of the children with abnormal newborn screening results for galactosemia and reduced GALT activities started treatment with dietary galactose restriction.
Sequence variations in the GALT gene in 13 Korean galactosemia patients
All 13 patients with reduced GALT activity had various sequence variations in the GALT gene. A total of 20 variant alleles (76.9%) were identified, corresponding to 12 different variations. We identified seven alleles with five known pathogenic mutant alleles, seven alleles with six novel likely pathogenic variant alleles, and six alleles with the Duarte variant allele. The five known mutations included four missense mutations [c.346C > A (p.Leu116Ile), c.602G > A (p.Arg201His), c.998G > A (p.Arg333Gln), and c.1087G > A (p.Glu363Lys)] and one intronic mutation (c.821-7A > G), which causes a splicing aberration (exon 9 deletion). The six novel likely pathogenic variations consisted of three missense variations [c.302C > A (p.Ala101Asp), c.493 T > C (p.Tyr165His), and c.769C > A (p.Pro257Thr)], one small deletion/insertion [c.826_827delinsAA (p.Ala276Asn)], one frameshift [c.286_299delGACAACGACTTCCC (p.Asp96Serfs*5)], and one splicing (c.378-1G > C) variation. Five patients had the Duarte variant allele (c.940A > G) and one was homozygous. Individual characteristics of the GALT genotype in the 13 patients used in this study are summarized in Table 1.
Structural and functional effects of novel variations
In silico analyses for four novel nucleotide substitutions and one novel in frame deletion/insertion mutation were predicted using PolyPhen-2 and SIFT. The possible mis-splicing effect of exonic or intronic variations predicted using Human Splice Finder is summarized in Table 2. Three novel variations of c.302C > A (p.Ala101Asp), c.796C > A (p.Pro257Thr), and c.826_827delinsAA (p.Ala276Asn) were predicted as mutations by PolyPhen-2 and SIFT. The novel variation of c.493 T > C (p.Tyr165His) was predicted to be a polymorphism by both PolyPhen-2 and SIFT. However, it was predicted to activate a cryptic splice site of an enhancer motif causing aberrant splicing according to the Human Splice Finder matrix. The c.493 T > C variation was observed in one patient who was a compound heterozygote for the previously reported pathogenic mutation, c.998G > A (p.Arg333Gln), and the patient had 6.3 μmol/h/g Hb of GALT activity, suggesting that this variation might be pathogenic.
The possible mis-splicing effect of silent exonic or intronic variations located outside of the canonical splicing signals, AG or GT, also revealed that c.302C > A (p.Ala101Asp) was predicted to activate a cryptic splice site of an enhancer motif. This variation was detected in one patient who was a compound heterozygote for the Duarte variant (D/G galactosemia) with 6.2 μmol/h/g Hb of GALT activity, which supports the hypothesis that this variation may be pathogenic rather than benign. The intronic variation of c.378-1G > C was predicted to impact the splice site through both activation of a cryptic splice site of an enhancer motif and disruption of a natural silencer motif. This variation was identified in one patient heterozygous for the other novel likely pathogenic variant allele [c.286_299delGACAACGACTTCCC (p. Asp96Serfs*5)] with GALT enzymatic activity below the quantitation limit of 0.1 μmol/h/g Hb. Also, another patient with 7.3 μmol/h/g Hb of GALT activity, was compound heterozygous for c.286_299delGACAACGACTTCCC and c.821-7A > G. A known mutant allele of c.821-7A > G was reported to cause an exon 9 deletion. In addition, six novel variations were absent in the 192 alleles from healthy individuals.
An intronic variation of c.82 + 20_82 + 60del was observed in one patient who was compound heterozygous for two other novel likely pathogenic variant alleles, c.286_299delGACAACGACTTCCC and c.378-1G > C, and had no GALT enzyme activity (<0.1 μmol/h/g Hb), which suggests that this variation is a polymorphism without significant enzymatic effect.
Mutation spectrum of the GALT gene in Korean patients
Table 3 summarizes all the variations in the GALT gene, including previously published data in Korea and this study. Thirty-four Korean patients with GALT deficiency galactosemia were recruited and a total of 51 variant alleles were identified (75.0%, 51/68 alleles). The most frequent nucleotide substitution was the Duarte variant (c.940A > G, n = 18, 35.3%), followed by c.507G > C (p.Gln169His, n = 5, 9.8%). Among the 34 Korean patients, eight (23.5%) had classic galactosemia with two pathogenic variant alleles, eight (23.5%) were compound heterozygous for a Duarte variant and a pathogenic variant allele, nine (26.5%) were heterozygous for only one pathogenic variant allele, eight (23.5%) were heterozygous for one Duarte variant allele, and one (2.9%) was homozygous for the Duarte variant allele. The GALT mutational spectrum is comprised of 18 distinct variations that are widely scattered. Most of the putative disease-causing variations were missense variations (n = 11, 61.1%), with the other variants being small deletion/insertions (n = 2, 11.1%), exonic or intronic variations that could affect splicing (n = 4, 22.2%) and silent exonic variations (n = 1, 5.6%).
GALT activities of all cases including our data are summarized in Figure 1. The GALT enzyme activities of patients varied according to genotype with overlapping distributions: mean 3.9 μmol/h/g Hb for classic galactosemia patients (range, < 0.1–7.3), mean 6.1 μmol/h/g Hb for compound heterozygotes of Duarte and classic variants (range, 5.9–6.2), 12.5 μmol/h/g Hb for Duarte homozygotes, mean 14.5 μmol/h/g Hb for carriers of classic mutant alleles (range, 11.6–16.4), and mean 14.4 μmol/h/g Hb for carriers of Duarte variant alleles (range, 12.6–16.1). GALT activity of classic galactosemia patients was significantly different from other groups with different variant alleles (p < 0.05). The differences of GALT activities between carriers of classic variant alleles and carriers of the Duarte variant allele were not significantly different (p > 0.05). Because only one patient was homozygous for the Duarte variant allele, it was difficult to compare GALT activities among different groups with variant alleles and there were no significant differences from other groups of patients with variant alleles except for the group of classic galactosemia patients (p > 0.05).

GALT enzyme activities among different categories of GALT biochemical phenotypes in 34 Korean patients. The bars in the boxes represent the median values, and the areas above and below the bars in the boxes represent the upper and lower quartiles, respectively. The lines extending below and above the boxes represent the lowest and highest values, respectively, and the circles represent individual data. P value was calculated by the comparison among five different groups of patients with GALT biochemical phenotypes by the Kruskal-Wallis test with post-hoc analysis (D, Duarte variant; G, mutant allele for galactosemia including novel likely pathogenic variant; N, normal allele). It is of note that although there were differences in GALT activity between the G/G and D/G patient groups, the GALT enzyme activities of individual patients were varied with overlapping distributions.
Discussion
Among 34 Korean patients with GALT mutations including likely pathogenic variants, the most common type of disease-causing mutation was a missense mutation (61.1%) followed by a splicing mutation (22.2%). The most common mutation except for the Duarte variant was c.507G > C (p.Gln169His) and it was found in five out of 51 variant alleles (9.8%). Other mutations previously found in the Korean population were widely scattered in our patients and no other specific mutations were found to be prevalent at a significant frequency, which makes targeted mutation analysis difficult.
Our study also reaffirmed that measurement of GALT enzyme activity was not enough to predict the genotypes and to make a decision for treatment in some patients because of the lack of correlation between genotype and phenotype [17],[18]. For the galactosemia variant, the D/G phenotype of GALT activity in which at least one mutation is the c.940A > G (p.Asn314Asp) allele with the enzyme activity between 5% and 50% of normal control values and with which there is no evidence for hepatocellular damage, cataract, or developmental delays during infancy, the need to restrict dietary galactose is uncertain [19],[20]. In our study, there were eight patients with the D/G GALT biochemical phenotype among 34 GALT-deficient galactosemia patients, and these patients had an enzyme activity of 5.9–15.8 μmol/h/g Hb (median 7.0 μmol/h/g Hb), corresponding to 21.5–57.5% of the mean control value. Although there were differences in GALT activity between the G/G and D/G patient groups, the GALT enzyme activities of individual patients were varied with overlapping distributions. It is reasonable and important to distinguish D/G galactosemia from G/G classic galactosemia using a molecular diagnostic test prior to making any treatment decisions. We report the first Korean patient with two Duarte variants who is in the homozygous state of the Duarte variant allele (D/D). Her GALT activity was 12.5 μmol/h/g Hb corresponding to 35.7–62.5% of normal control values (20–35 μmol/h/g Hb), which is consistent with previously reported data [21].
The incidence of GALT galactosemia varies, ranging from 1 in 23,000–40,000 in Europe [17],[22] to 1 in 50,000 in the United States [10]. It has been reported that the frequency of GALT galactosemia has decreased in continental Europe, moving through populations in an eastern and southern direction [17]. Using haplotype analysis in conjunction with a measure of genetic diversity and linkage disequilibrium, it was found that p.Gln188Arg arose in central Europe within the last 20,000 years, possibly due to population expansion during the re-colonization of Europe by Homo sapiens in the Mesolithic Age. p.Lys285Asn is a younger mutation that originated in Eastern Europe and is probably more geographically restricted since it arose after all major European population expansions [23]. The Duarte variant, c.940A > G (p.Asn314Asp), was found to be an ancient mutation that originated before the expansion of Homo sapiens out of Africa [10],[23]. In Asian countries, the incidences of classic galactosemia is reported to be much lower than in Western countries, ranging from 1 in 1,000,000 in Japan to 1 in 500,000 in China [6].
In Korea, the nationwide newborn screening for galactosemia was first introduced in 1991. From 1997 to 2005, it was stopped due to budget concerns and cost-effectiveness of the newborn screening program and then resumed in 2006 [24]. According to the annual report of the Ministry of Health and Welfare of Korea, about 80.4% of newborns were screened by the nationwide newborn screening program from 1991 to 2007. After 2006, the incidence of galactosemia has increased from 1:100,000 in 2006 to around 1:15,000 in 2012, and prevalence of galactosemia in Korea was calculated to be between 1:30,000 and 1:35,000 from 2005 to 2012 [13],[25]. However, the exact incidence and prevalence of classic galactosemia is not yet known. Although it is very difficult to evaluate the accurate prevalence of classic galactosemia in the Korean population, some previous reports have suggested a lower prevalence in Koreans compared to Western countries [13]-[15],[24]. Ko et al. reported only one classic galactosemia patient in 18 patients with reduced GALT activities [14], and Lee et al. reported three classic galactosemia patients during the 11-year study period [15], which suggested a low prevalence of classic galactosemia in the Korean population. However, during the 3-year period in this study, which was relatively shorter than in previous studies, four classic galactosemia patients were diagnosed, which suggests that the prevalence of classic galactosemia in the Korean population may be increasing. One cause for the increase in galactosemia incidence could be due to a different panel of populations screened. Another reason attributing to the higher prevalence observed may be due to different methods used more recently with respect to old methods. Lastly, other unknown variables that have been taken into account to explain the increasing of prevalence. In Korea, the accreditation and inspection program by the Planned Population Federation of Korea for the proper quality assurance of clinical laboratory performing newborn screening tests has been settled since 2008 which could also affect the increasing prevalence of galactosemia [4],[25]. Further studies are needed to evaluate the accurate prevalence of classic galactosemia combined with comprehensive monitoring program.
To date, more than 230 mutations have been identified in the GALT gene databases [8],[9]. Eight common GALT mutations for targeted mutation analysis of p.Gln188Arg, p.Ser135Leu, p.Lys285Asn, p.Leu195Pro, p.Tyr209Cys, p.Phe171Ser, c.253-2A > G, and a 5 kb deletion have been introduced as a cost-effective diagnostic strategy in Western countries [10]-[12]. Also, other diagnostic methods such as matrix assisted laser desorption/ionization time-of-flight mass spectrometry, PCR using an amplification refractory mutation system, and multiplexed single nucleotide extension techniques have also been introduced for both population-based screening and diagnosis in patients [4],[18],[26]-[28]. However, none of the above eight common mutations have been found in the Korean population as well as another East Asian populations including Chinese and Japanese [6],[7],[14],[15],[29]. These findings strongly indicate that the Asian GALT mutations are clearly different from the European mutations because of the genetic divergence of Homo sapiens[6],[17],[23]. Although further evaluation is needed to confirm the origin and the exact effect of the six novel likely pathogenic variations identified in this study, our work suggests that these variants could be young mutations that may only be found in those of Asian ethnicity. Taking into account of all these findings, it is reasonable to suggest that the mutation databases in the Asian population be separated or regarded as a distinct category from those of Western countries for the best diagnostic approach and for development and application of therapeutic approaches in Asian patients with GALT-deficient galactosemia.
It is possible that the difference in the prevalence of classic galactosemia between countries is due to technical issues with the population-based newborn screening programs rather than true population differences [4]. Differences in approach or sensitivity of the methods used to detect GALT deficiency, or in defined cut-off levels, may leave some heterozygotes and/or Duarte variants included in some population estimates but not others [4]. For example, Africa has an estimated prevalence of 1:14,000, which is based on a population-based screen of cord blood samples for the p.Ser135Leu GALT mutation and accounts for > 90% of all classic galactosemia cases in this population [30]. The increasing detection rate of galactosemia in the Korean population should be further evaluated with a focus on the screening procedures.
Conclusions
In summary, we identified six novel likely pathogenic variations and expanded the genetic spectrum of GALT variations in Koreans. The mutations responsible for GALT deficiency in the Korean population were clearly different from those of other populations, and common mutations with high frequency in Western countries are not found in Koreans. GALT mutations are very widely scattered along the entire length of the gene, so targeted mutational analysis for screening or diagnosis is not appropriate.
Compliance with ethics guideline
The content of this article represents original work that has not been previously published nor in consideration of publication elsewhere. The manuscript has been read and approved for submission to the journal by all contributing authors.
References
Leslie ND, Immerman EB, Flach JE, Florez M, Fridovich-Keil JL, Elsas LJ: The human galactose-1-phosphate uridyltransferase gene. Genomics. 1992, 14 (2): 474-480. 10.1016/S0888-7543(05)80244-7.
Bosch AM: Classical galactosaemia revisited. J Inherit Metab Dis. 2006, 29 (4): 516-525. 10.1007/s10545-006-0382-0.
Waisbren SE, Potter NL, Gordon CM, Green RC, Greenstein P, Gubbels CS, Rubio-Gozalbo E, Schomer D, Welt C, Anastasoaie V, D’Anna K, Gentile J, Guo CY, Hecht L, Jackson R, Jansma BM, Li Y, Lip V, Miller DT, Murray M, Power L, Quinn N, Rohr F, Shen Y, Skinder-Meredith A, Timmers I, Tunick R, Wessel A, Wu BL, Levy H: The adult galactosemic phenotype. J Inherit Metab Dis. 2012, 35 (2): 279-286. 10.1007/s10545-011-9372-y.
Jumbo-Lucioni PP, Garber K, Kiel J, Baric I, Berry GT, Bosch A, Burlina A, Chiesa A, Pico ML, Estrada SC, Henderson H, Leslie N, Longo N, Morris AA, Ramirez-Farias C, Schweitzer-Krantz S, Silao CL, Vela-Amieva M, Waisbren S, Fridovich-Keil JL: Diversity of approaches to classic galactosemia around the world: a comparison of diagnosis, intervention, and outcomes. J Inherit Metab Dis. 2012, 35 (6): 1037-1049. 10.1007/s10545-012-9477-y.
Bosch AM, Ijlst L, Oostheim W, Mulders J, Bakker HD, Wijburg FA, Wanders RJ, Waterham HR: Identification of novel mutations in classical galactosemia. Hum Mutat. 2005, 25 (5): 502-10.1002/humu.9330.
Hirokawa H, Okano Y, Asada M, Fujimoto A, Suyama I, Isshiki G: Molecular basis for phenotypic heterogeneity in galactosaemia: prediction of clinical phenotype from genotype in Japanese patients. Eur J Hum Genet. 1999, 7 (7): 757-764. 10.1038/sj.ejhg.5200368.
Cheung KL, Tang NL, Hsiao KJ, Law LK, Wong W, Ng PC, Pang CP, Applegarth DA, Fok TF, Hjelm NM: Classical galactosaemia in Chinese: a case report and review of disease incidence. J Paediatr Child Health. 1999, 35 (4): 399-400. 10.1046/j.1440-1754.1999.00373.x.
Calderon FR, Phansalkar AR, Crockett DK, Miller M, Mao R: Mutation database for the galactose-1-phosphate uridyltransferase (GALT) gene. Hum Mutat. 2007, 28 (10): 939-943. 10.1002/humu.20544.
The human gene mutation database.., [http://www.hgmd.org/]
Suzuki M, West C, Beutler E: Large-scale molecular screening for galactosemia alleles in a pan-ethnic population. Hum Genet. 2001, 109 (2): 210-215. 10.1007/s004390100552.
Elsas LJ, Lai K: The molecular biology of galactosemia. Genet Med. 1998, 1 (1): 40-48. 10.1097/00125817-199811000-00009.
Coffee B, Hjelm LN, DeLorenzo A, Courtney EM, Yu C, Muralidharan K: Characterization of an unusual deletion of the galactose-1-phosphate uridyl transferase (GALT) gene. Genet Med. 2006, 8 (10): 635-640. 10.1097/01.gim.0000237720.78475.fb.
Lee DH: The prevalence of pediatric endocrine and metabolic diseases in Korea. Korean J Pediatr. 2008, 51 (6): 559-563. 10.3345/kjp.2008.51.6.559.
Ko DH, Chang HE, Song SH, Park KU, Kim JQ, Kim MC, Song YH, Hong YH, Lee DH, Song J: Molecular and biochemical characterization of the GALT gene in Korean patients with galactose-1-phosphate uridyltransferase deficiency. Clin Chim Acta. 2010, 411 (19–20): 1506-1510. 10.1016/j.cca.2010.06.008.
Lee BH, Cheon CK, Kim JM, Kang M, Kim JH, Yang SH, Kim GH, Choi JH, Yoo HW: Low prevalence of classical galactosemia in Korean population. J Hum Genet. 2011, 56 (1): 94-96. 10.1038/jhg.2010.152.
Shin YS: Galactose metabolites and disorders of galactose metabolism. Techniques in Diagnostic Human Biochemical Genetics. Edited by: Hommes FA. 1991, Wiley-Liss, New York, 267-283.
Tyfield L, Reichardt J, Fridovich-Keil J, Croke DT, Elsas LJ, Strobl W, Kozak L, Coskun T, Novelli G, Okano Y, Zekanowski C, Shin Y, Boleda MD: Classical galactosemia and mutations at the galactose-1-phosphate uridyl transferase (GALT) gene. Hum Mutat. 1999, 13 (6): 417-430. 10.1002/(SICI)1098-1004(1999)13:6<417::AID-HUMU1>3.0.CO;2-0.
Jama M, Nelson L, Pont-Kingdon G, Mao R, Lyon E: Simultaneous amplification, detection, and analysis of common mutations in the galactose-1-phosphate uridyl transferase gene. J Mol Diagn. 2007, 9 (5): 618-623. 10.2353/jmoldx.2007.070027.
Ficicioglu C, Thomas N, Yager C, Gallagher PR, Hussa C, Mattie A, Day-Salvatore DL, Forbes BJ: Duarte (DG) galactosemia: a pilot study of biochemical and neurodevelopmental assessment in children detected by newborn screening. Mol Genet Metab. 2008, 95 (4): 206-212. 10.1016/j.ymgme.2008.09.005.
Fernhoff PM: Duarte galactosemia: how sweet is it?. Clin Chem. 2010, 56 (7): 1045-1046. 10.1373/clinchem.2010.147371.
Lai K, Langley SD, Dembure PP, Hjelm LN, Elsas LJ: Duarte allele impairs biostability of galactose-1-phosphate uridyltransferase in human lymphoblasts. Hum Mutat. 1998, 11 (1): 28-38. 10.1002/(SICI)1098-1004(1998)11:1<28::AID-HUMU5>3.0.CO;2-H.
Murphy M, McHugh B, Tighe O, Mayne P, O’Neill C, Naughten E, Croke DT: Genetic basis of transferase-deficient galactosaemia in Ireland and the population history of the Irish Travellers. Eur J Hum Genet. 1999, 7 (5): 549-554. 10.1038/sj.ejhg.5200327.
Flanagan JM, McMahon G, Brendan Chia SH, Fitzpatrick P, Tighe O, O’Neill C, Briones P, Gort L, Kozak L, Magee A, Naughten E, Radomyska B, Schwartz M, Shin JS, Strobl WM, Tyfield LA, Waterham HR, Russell H, Bertorelle G, Reichardt JK, Mayne PD, Croke DT: The role of human demographic history in determining the distribution and frequency of transferase-deficient galactosaemia mutations. Heredity. 2010, 104 (2): 148-154. 10.1038/hdy.2009.84.
Lee DH: Newborn screening of inherited metabolic disease in Korea. Korean J Pediatr. 2006, 49 (11): 1125-1139. 10.3345/kjp.2006.49.11.1125.
Analysis of Blood Sample Records for Neonatal Screening Test, External Quality Assessment and Field Assessment for Inborn Errors of Metabolism in Korea. 2012, Welfare MoH, Seoul, Korea
Goldstein N, Cohen Y, Pode-Shakked B, Sigalov E, Vilensky B, Peleg L, Anikster Y: The GALT rush: high carrier frequency of an unusual deletion mutation of the GALT gene in the Ashkenazi population. Mol Genet Metabol. 2011, 102 (2): 157-160. 10.1016/j.ymgme.2010.10.007.
Barbouth D, Slepak T, Klapper H, Lai K, Elsas LJ: Prevention of a molecular misdiagnosis in galactosemia. Genet Med. 2006, 8 (3): 178-182. 10.1097/01.gim.0000204019.54509.40.
Mahmood U, Imran M, Naik SI, Cheema HA, Saeed A, Arshad M, Mahmood S: Detection of common mutations in the GALT gene through ARMS. Gene. 2012, 509 (2): 291-294. 10.1016/j.gene.2012.08.010.
Ashino J, Okano Y, Suyama I, Yamazaki T, Yoshino M, Furuyama J, Lin HC, Reichardt JK, Isshiki G: Molecular characterization of galactosemia (type 1) mutations in Japanese. Hum Mutat. 1995, 6 (1): 36-43. 10.1002/humu.1380060108.
Henderson H, Leisegang F, Brown R, Eley B: The clinical and molecular spectrum of galactosemia in patients from the Cape Town region of South Africa. BMC Pediatr. 2002, 2: 7-10.1186/1471-2431-2-7.
Acknowledgements
This study was supported by a grant from the Korea Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (A120030).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RC analyzed genetic data and was responsible for drafting the manuscript. KJ investigated clinical and biochemical information of the patients. DK analyzed biochemical and molecular genetic data. DL was supplier of samples and clinical data. JS was supplier of samples and clinical data. DJ was supplier of samples and clinical data. CK carried out the PCR and sequencing analysis. SL carried out the biochemical analysis. JK helped to draft the manuscript. YL revised the manuscript for important intellectual content. HP coordinated whole research and helped to revise the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
Cite this article
Choi, R., Jo, K.I., Ko, DH. et al. Novel GALTvariations and mutation spectrum in the Korean population with decreased galactose-1-phosphate uridyltransferase activity. BMC Med Genet 15, 94 (2014). https://doi.org/10.1186/s12881-014-0094-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-014-0094-5