
Abstract
Background
In recent decades, Mycobacterium tuberculosis with the RDRio genotype, frequently isolated from tuberculosis patients in Rio de Janeiro, has become part of the Latin American – Mediterranean (LAM) family and has been associated with multidrug-resistant tuberculosis (MDR-TB). The aim of this study was to investigate the frequency of M. tuberculosis RDRio in the state of Minas Gerais, Brazil, and its relationship with MDR-TB.
Methods
For convenience, 172 susceptible and 63 MDR M. tuberculosis isolates were taken from pulmonary samples from patients diagnosed between January 2007 and December 2011. The DNA extracted from these isolates was analyzed by spoligotyping, PCR-RFLP to characterize fbpC103/Ag85C103, multiplex PCR to detect RDRio and RD174, and MIRU-VNTR 24 loci.
Results
Among the 235 isolates, the RDRio pattern was identified in 122 (51.9%) isolates (IC 0.45–0.58), with 100 (42.5%) wild-type and 13 (5.5%) mixed pattern isolates, whereas RD174 was identified in 93 of the 122 RDRio positive samples (76.3%). The LAM family and the LAM9 lineage were the most frequently identified among the isolates in this study. Among the 63 MDR isolates, 41 (65.1%) were RDRio and 28 (44.4%) RD174.
Conclusion
The association of both deletions with MDR proved to be statistically significant, corroborating the few reports that have associated RDRio with MDR.
Similar content being viewed by others

Background
In 2017, 10 million new cases of tuberculosis (TB) were reported worldwide, with 558,000 new cases of rifampicin-resistant tuberculosis (RR-TB). Among RR-TB cases, an estimated 82% had multidrug-resistant TB (MDR-TB), and in Brazil in the same year, there were 2000 cases of MDR/RR-TB among pulmonary TB cases [1]. Mycobacterium tuberculosis (M. tuberculosis) is a human pathogen that undergoes clonal evolution, resulting in divergent lineages associated with specific geographic regions, and possibly with different human ethnic populations [2]. These varied lineages present with biological differences regarding transmissibility [3].
Molecular analyses based on specific genetic markers enable the rapid identification of different species and sublineages, an important tool for studying the evolution and transmission of M. tuberculosis [4]. The marker used to characterize the Latin American - Mediterranean (LAM) family is the single nucleotide polymorphism (SNP) fbpC103/Ag85C103, which is considered important for identification due to its high specificity. In addition, regions of difference such as RDRio and RD174 are also lineage specific, and the latter has been associated with higher levels of transmissibility [4].
The LAM family accounts for approximately 15% of the global burden of TB, and is present in 46% of the isolates that have been analyzed through genotyping in Brazil [4]. The LAM9 lineage in particular represents 10.2% of the isolates of M. tuberculosis on the American continent [5]. In 2007, Lazzarini et al. [6] described a genotype of M. tuberculosis called RDRio, which is exclusively found as a sublineage derived from the LAM family [6]. It is believed that this genotype originated from a progenitor LAM9 and gave rise to the sublineages LAM1, LAM2, LAM4, and LAM5, as documented by the successive loss of spacers in spoligotype patterns [6,7,8].
The M. tuberculosis RDRio genotype contains a 26.3 kb deletion that causes the loss and modification of 10 genes, including two PPE (Proline-glutamic Acid Proteins) genes which encode specific proteins important for immunomodulation [5]. This sublineage has been isolated in several places in Brazil and other countries, and has been associated with higher levels of transmission, such as that found in MDR-TB [2, 3, 7].
Another important molecular technique that allows for the phylogenetic study of the M. tuberculosis complex is the Variable Number Tandem Repeat – Mycobaterial Interspersed Repetitive Unit (MIRU-VNTR 24 loci) based genotyping tool. This, together with spoligotyping, resulted in the construction of large genotypic databases that allowed for the phylogenetic analysis and study on the global distribution of M. tuberculosis [9]. Genotyping by means of MIRU-VNTR 24 loci also enables the study of the epidemiologically significant clonal diversity of M. tuberculosis strains, which is useful for exploring internal phylogenetic ramifications [7, 9,10,11]. There are some studies that describe the frequency of M. tuberculosis RDRio in certain populations, but data evaluating the significance of this relationship by means of specific statistical tests on the co-occurrence of this genotype with MDR-TB are scarce, and no such data is available for the state of Minas Gerais [2, 3, 12]. In this context, the aim of this study was to evaluate the frequency of M. tuberculosis RDRio isolation in this region of Brazil, and its relationship with MDR-TB and other genetic markers affecting the drug susceptibility profile.
Methods
Study design
For convenience, 172 susceptible and 63 MDR-TB (strains defined as drug resistant to at least isoniazid and rifampin) M. tuberculosis isolates were collected. Isolates with single-drug resistance were not included.
The isolates were obtained from pulmonary samples (sputum and bronchoalveolar lavage) from patients diagnosed between January 2007 and December 2011, in Minas Gerais.
In this state, the mean MDR-TB rate was 0.2% among clinical TB cases between 2002 and 2009 [13], while after this period MDR-TB detection rates increased due to the expansion of culture and the susceptibility testing. In the present study, the identification of M. tuberculosis was performed by phenotypic testing [14], while drug susceptibility testing was conducted using the BACTEC™ MGIT™ 960 system (Becton Dickinson®), according to manufacturer’s instructions [15], in the Main Public Health Laboratory of the state of Minas Gerais located in the Octavio Magalhães Institute of the Ezequiel Dias Foundation in Belo Horizonte. All 235 clinical isolates were analyzed by means of the molecular tests described below, the results of which are included in the statistical analyses.
DNA extraction
The genomic DNA of M. tuberculosis was extracted from subcultured colonies in Lowenstein-Jensen solid medium using 10% Cetyltrimethylammonium Bromide (CTAB) as described by Dantas et al. [16] in 2015. The extracted DNA was used for the techniques described below. The experiments were performed in duplicate, with the exception of spoligotyping and MIRU-VNTR 24 loci.
Multiplex PCR - RDRio
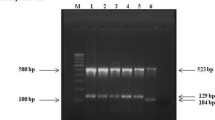
The detection of the RDRio pattern was performed by multiplex PCR, using the following set of primers: BridgeRDRioF, BridgeRDRioR, IS1561F, and IS161R, and was confirmed by the presence or absence of 1175 and/or 530 bp fragments. The RDRio pattern presents a 1175 bp fragment, while the wild-type (WT) produces a 530 bp fragment [4, 6, 7].
Multiplex PCR - RD174
To perform amplification, we used the PCR protocol describe by Gibson et al. [4] in 2008, and adapted by Vasconcelos et al. [7] in 2014. Briefly, we used a primer for each of the following: RD174 F, RD174Fi, and RD174 R. The isolates showing an intact RD174 region (WT) produced 300 bp fragments, while the RD174 deletion presented as 500 bp fragments [4, 7].
Spoligotyping
Spoligotyping was performed using the Beamedex® microsphere technique in a Luminex-Bioplex-BioRad 200® system, as developed in the “Institut de Genétique et Microbiologi e Université Paris-Sud”, and following the protocol described by Zhang et al. (2010) [17].
PCR-RFLP fbpC103/Ag85C103
The SNP fbpC103,or Ag85C103, was described as a specific marker for the LAM lineage by Gibson et al. [4] in 2008. The PCR protocol used in this study was adapted as described by Vasconcelos et al. [7] in 2014. To perform the amplification, we used this set of primers: fbpC103 F and fbpC103 R. The amplified products (519 bp) were analyzed on 2% agarose gel in 1× TBE. After this step, the enzymatic digestion was performed by restriction enzyme MnII (New England BioLabs Inc. USA). The MnII enzyme produces three restriction fragments in the amplified product: 365 bp, 96 bp, and 48 bp. The presence of SNP (G309A) (LAM) results in the loss of one of the three restriction sites [4, 7].
MIRU-VNTR 24 loci
The MIRU-VNTR 24 loci was performed according to the protocol described by Supply et al. [9] in 2006. This procedure used a monoplex PCR, using the fragments revealed by electrophoresis, in 2% agarose gel. In the construction of the dendogram, the Neighbor-Joining (NJ) algorithm was used to analyze the categorical data.
Statistical analyses
Associations were calculated using the chi-square and Fisher’s tests, and performed by STATA 12 software (Copyright 1985–2015 StataCorpLP©, USA).
Phylogenetic analyses
The analyses were performed using these free sites: the SITVIT website http://www.pasteur-guadeloupe.fr:8081/SITVIT_ONLINE/ and the MIRU-VNTRplus Server Policy http://www.miru-vntrplus.org/MIRU/index.faces , as well as with Bionumerics 7.0©Applied Math software (Sint maertens, Latem, Belgium).
Results
Mycobacterium tuberculosis RDRio - frequency and association with MDR-TB
Among the 235 M. tuberculosis isolates, 122 (51.9%) were identified as RDRio (IC 0.45–0.58), 100 (42.5%) as NO-RDRio, and 13 (5.5%) as mixed pattern. The relationship between the RDRio sublineage and MDR-TB was significant (p < 0.001, chi-square test). The percentages of RDRio and NO-RDRio in susceptible and MDR-TB clinical isolates are shown in Fig. 1.

Mycobacterium tuberculosis RDRio and its association with MDR-TB. This grafics shows the percentages of RDRio and NO-RDRio in susceptible and MDR-TB clinical isolates. It’s important to highlight the relationship between the RDRio sublineage and MDR-TB was significant (p < 0.001)
Of the 122 M. tuberculosis RDRio, 116 (95.1%) were identified as LAM, while the remaining six (4.9%) were identified as WT based on the Ag85C103 SNP, and this relationship was significant (p < 0.000, chi-square test, and p < 0.000, Fisher’s exact test). The genetic profiles of these six RDRio, classified as WT by means of AG85C103 SNP, and found after analyses performed by spoligotyping and MIRU-VNTR 24 loci, are described in Table 1.
The spoligotyping show that the majority of the RDRio isolates proved to be LAM. The RDRio spoligotyping’s patterns are shown in Fig. 2. The distribution of profiles found among drug resistant and susceptible isolates is shown in Additional file 1. The spoligotyping classifications for all 172 sensitive and 63 MDR isolates are described in Table 2.

The spoligotyping of M. tuberculosis RDRio. A = The distribuition of RDRio spoligotyping’s pattern of the LAM family. B = The distribuition of RDRio spoligotyping’s pattern of the NO-LAM families
Identification of the RD174 pattern
Among the 235 isolates of M. tuberculosis, the RD174 pattern was identified in 98 (41.7%), while 111 (47.2%) were WT and 26 (11.1%) presented a mixed pattern (RD174/WT).
Of the 98 isolates that presented as RD174, 93/98 (94.9%) were identified as RDRio (p < 0.000, chi-square test).
Among the 63 resistant M. tuberculosis, 28 (44.4%) presented the RD174 pattern, while among the 172 sensitive isolates 90 (52.3%) presented the WT pattern. The relationship between the RD174 pattern and TB drug resistance was significant, as was relationship between the WT pattern and drug sensitivity (p < 0.001, chi-square test, and p < 0.002, Fisher’s exact test).
Identification of the Ag85C103 SNP and comparison with spoligotyping
Of the 235 M. tuberculosis identified by the Ag85C103 SNP, 175 (74.4%) were classified as LAM, 54 (23%) as non-LAM, and six (2.5%) presented a mixed pattern (SNP/nonSNP). The profile of these isolates is described in Table 3, of which only one presented a mixed pattern concurrent with the other markers (RDRio and RD174). The relationship between these and the spoligotyping results is shown in Fig. 3.

The relationship between Ag85C103 SNP and spoligotyping. Purple circle – spoligotyping, Blue circle - Ag85C103
Analysis of the Ag85C103 SNP detected a higher frequency of LAM genotypes than spoligotyping, and this difference proved to be significant (p < 0.000, chi-square test, and p < 0.003, Fisher’s exact test).
No significant difference was observed between susceptible and resistant M. tuberculosis in isolates characterized as LAM by Ag85C103 (p < 0.309, chi-square test, and p < 0.428, Fisher’s exact test).
MIRU-VNTR 24 loci
Among the 235 isolates, MIRU-VNTR 24 loci typing revealed the following families: LAM, 136 (69.4%); Haarlem, 36 (15.3%); Cameroon, 13 (5.5%); X, 10 (4.3%); Unganda I, 7 (3%); Delhi/CAS, 4 (1.7%); one from Ghana (0.4%), and one from a NEW-1 type. A UPGMA-based dendrogram demonstrates the four clusters of the RDRio sublineage (Figs. 4 and 5).

Dendogram of association by MIRU-VNTR 24 loci, spoligotyping, and RD174. A = Dendogram with association of all strains with the pattern RD174 and this respective MIRU-VNTR 24 loci. B = The spoligotyping of all strains with the pattern RD174

Dendogram of association by MIRU-VNTR 24 loci, spoligotyping, and RDRio. A = Dendogram with association of all strains with the pattern RDRio and this respective MIRU-VNTR 24 loci. B = The spoligotyping of all strains with the pattern RDRio
Discussion
The present study demonstrates a significant relationship between the RDRio sublineage and MDR-TB, and is the first study of its kind in the state of Minas Gerais. In contrast to previous studies that only evaluated the frequency of this sublineage, it is important to note in the present study that we also evaluated the relationship of not only RDRio, but also of other phylogenetic markers (RD174, Ag85C103 SNP), with their susceptibility or resistance to anti-TB drugs.
Lazzarini et al. [6] in 2007 suggested that the M. tuberculosis genotype RDRio originated from a common progenitor, since IS1561 deletion was also found in other countries. In addition, besides their capacity for progressive primary TB, isolates with this genotype have been associated with the development of the MDR phenotype [4, 6, 7].
The high frequency of RDRio observed in this study population may well be due to the predominance of the LAM family and the LAM9 lineage, since they are the most common progenitors of this sublineage [3, 4, 6, 7]. We also observed a statistically significant predominance of the RDRio sublineage among MDR isolates, as compared to sensitive isolates. This result is consistent with data obtained in Porto Alegre, where RDRio was observed in 56 of the 115 MDR isolates, accounting for almost half of the resistant isolate cases [12]. In a study performed in Portugal, the RDRio frequency was 60% among MDR isolates [3], while in the United States and Spain the RDRio frequency in MDR isolates was no higher than in sensitive isolates, but was identified among both MDR and isoniazid monoresistant isolates [2, 12]. In those same studies in Spain and the United States, RDRio was found in a higher proportion in Hispanic patients [2, 18]. Lazzarini et al. [19] analyzed isolates from several parts of Brazil in 2008, and suggested that the sublineage RDRio LAM could cause more severe disease (cavitary lung lesions), and most likely contributes to the transmission of TB among certain ethnic populations [6, 12, 19].
RDRio may have some biological advantage over other genotypes due to the deletion of two PPE genes (PPE55 and PPE56), and may minimize the host’s immunological recognition leading to increased virulence and/or transmissibility [4, 12].
In a study that evaluated RDRio in TB contacts in Gambia [20], RD174 was the second most frequent marker found, suggesting a certain association with the RDRio deletion. The most frequent marker was RD702, but this was related to the transmission of M. africanum [20].
In our study, although RD174 was identified in most of the isolates with RDRio (94.9%), and though the relationship between these markers was significant (p = 0.000), their correlation is not absolute. Therefore, when analyzing only RD174, one can overestimate the RDRio frequency in the present population, as described by another author that analyzed isolates from Rio de Janeiro [7]. This data contradicts a study by Gibson et al. [4] in 2008, who considered RD174 an absolute marker for RDRio. One explanation for this difference may well be the local features of sample selection.
As shown in this work, spoligotyping exhibits limitations in differentiating Euro American families in populations that predominate with the LAM, H, and T families, and the fact that we found six RDRio isolates characterized as Non-LAM by spoligotyping may be due to these limitation [4, 7, 8]. The difficulty in differentiating these families comes from the large number of IS6110 of these strains produce, with many variations in the “Direct Repeat” (DR) locus, which gives rise to several profiles not identified by this technique (Unkonwn pattern), or with a high degree of homoplasy between the spacers that define different families [15, 16]. In this context, the Ag85C103 SNP is particularly important as a specific additional marker used to identify the frequency of the LAM family in a population [2, 4].
One limitation of this study is that since the RDRio sublineage could not be correlated with patients’ clinical data, it was impossible to associate RDRio strains with a prognosis for severe forms of TB. Another limitation is that we did not perform an entire genome analysis of the present strain’s population, and it has been shown that differences occur between this level of genotyping and 24 MIRU-VNTR typing and lineage definition. This finding would be particularly interesting for better characterizing the isolates that present with either RDRio or RD174, but not both. However, we must emphasize that the methods employed in this study are feasibly useful in places with few resources. In contrast, though WGS reduces the processing time, it is still an expensive technology that demands heavy bioinformatics to analyze the data. The results of this study were obtained through simple molecular biology techniques and may be important for the control and monitoring of TB and TBMDR.
Conclusions
In conclusion, most of the isolates of M. tuberculosis in Minas Gerais belong to the LAM family, the LAM9 lineage, and the RDRio sublineage. Both M. tuberculosis RDRio and RD174 isolates positively correlated with MDR-TB. Because Brazil has a vast territory with considerable demographic and economic differences, further studies of RDRio in other regions are required.
Availability of data and materials
The analyses for objectives of this study were conducted using STATA 12 software (Copyright 1985–2015 StataCorpLP©, USA). The data that support the findings of this study are available from Silvana Spindola de Miranda upon reasonable request by qualified researchers.
Abbreviations
- LAM:
-
Latin American – Mediterranean
- M. tuberculosis :
-
Mycobacterium tuberculosis
- MDR-TB:
-
Multidrug-resistant tuberculosis
- MIRU-VNTR:
-
Variable Number Tandem Repeat – Mycobaterial Interspersed Repetitive Unit
- PPE:
-
Proline-glutamic Acid Proteins
- RR-TB:
-
Rifampicin-resistant tuberculosis
- SNP:
-
Single Nucleotide Polymorphism
- TB:
-
Tuberculosis
- WT:
-
Wild-type
References
World Health Organization. Global tuberculosis report 2018. Geneva: World Health Organization; 2018.
Weisenberg SA, Gibson AL, Huard RC, Kurepina N, Bang H, Lazzarini LC, et al. Distinct clinical and epidemiological features of tuberculosis in new York City caused by the RDRio mycobacterium tuberculosis sublineage. Infect Genet Evol. 2014;27:380–92.
David S, Duarte EL, Leite CQF, Ribeiro JN, Maio JN, Paixão E, et al. Implication of the RDRio mycobacterium tuberculosis sublineage in multidrug resistant tuberculosis in Portugal. Infect Genet Evol. 2012;12:1362–7. https://doi.org/10.1016/j.meegid.2012.04.021.
Gibson AL, Huard RC, Gey Van Pittius NC, Lazzarini LCO, Driscoll J, Kurepina N, et al. Application of sensitive and specific molecular methods to uncover global dissemination of the major RDRio sublineage of the Latin American-Mediterranean mycobacterium tuberculosis spoligotype family. J Clin Microbiol. 2008;46:1259–67.
Reynaud Y, Millet J, Rastogi N. Genetic structuration, demography and evolutionary history of mycobacterium tuberculosis LAM9 sublineage in the Americas as two distinct subpopulations revealed by Bayesian analyses. PLoS One. 2015;10:1–15.
Lazzarini LCO, Huard RC, Boechat NL, Gomes HM, Oelemann MC, Kurepina N, et al. Discovery of a novel mycobacterium tuberculosis lineage that is a major cause of tuberculosis in Rio de Janeiro, Brazil. J Clin Microbiol. 2007;45:3891–902.
Vasconcellos SEG, Acosta CC, Gomes LL, Conceição EC, Lima KV, De Araujo MI, et al. Strain classification of mycobacterium tuberculosis isolates in Brazil based on genotypes obtained by spoligotyping, mycobacterial interspersed repetitive unit typing and the presence of large sequence and single nucleotide polymorphism. PLoS One. 2014;9.
Gomes HM, Elias AR, Oelemann MAC, Pereira MA d S, Montes FFO, Marsico AG, et al. Spoligotypes of mycobacterium tuberculosis complex isolates from patients residents of 11 states of Brazil. Infect Genet Evol. 2012;12:649–56. https://doi.org/10.1016/j.meegid.2011.08.027.
Supply P, Allix C, Lesjean S, Cardoso-Oelemann M, Rüsch-Gerdes S, Willery E, et al. Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing of mycobacterium tuberculosis. J Clin Microbiol. 2006;44:4498–510.
Cardoso Oelemann M, Gomes HM, Willery E, Possuelo L, Batista Lima KV, Allix-Béguec C, et al. The forest behind the tree: phylogenetic exploration of a dominant mycobacterium tuberculosis strain lineage from a high tuberculosis burden country. PLoS One. 2011;6.
Augusto CJ, Carvalho WDS, Almeida IND, Figueiredo LJDA, Dantas NGT, Suffys PN, et al. Comparative study of RFLP-IS6110 and MIRU-VNTR from mycobacterium tuberculosis isolated in the state of Minas Gerais, Brazil. Brazilian J Microbiol. 2018;49.
Dalla Costa ER, Ribeiro MO, Silva MS, Arnold LS, Rostirolla DC, Cafrune PI, et al. Correlations of mutations in katG, oxyR-ahpC and inhA genes and in vitro susceptibility in mycobacterium tuberculosis clinical strains segregated by spoligotype families from tuberculosis prevalent countries in South America. BMC Microbiol. 2009;9.
Augusto CJ, Carvalho WS, Gonçalves AD, Ceccato MGB, de Miranda SS. Characteristics of tuberculosis in the state of Minas Gerais, Brazil: 2002-2009. J Bras Pneumol. 2013;39.
BRASIL. Manual nacional de vigilância laboratorial da tuberculose e outras micobactérias. Secretaria de Vigilância em Saúde, Departamento de Vigilância Epidemiológica - Ministério da Saúde 2011.
Siddiqi SH, Rüsch-Gerdes S. For BACTEC™ MGIT 960™ TB System (Also applicable for Manual MGIT) 2006; July.
Dantas NGT, Suffys PN, Carvalho WS, Gomes HM, de Almeida IN, de Assis LJ, et al. Genetic diversity and molecular epidemiology of multidrug-resistant mycobacterium tuberculosis in Minas Gerais state, Brazil. BMC Infect Dis. 2015;15.
Zhang J, Abadia E, Refregier G, Tafaj S, Boschiroli ML, Guillard B, et al. Mycobacterium tuberculosis complex CRISPR genotyping: improving efficiency, throughput and discriminative power of “spoligotyping” with new spacers and a microbead-based hybridization assay. J Med Microbiol. 2010;59:285–94.
Ippolito G, Nisii C, Di Caro A, Brown D, Gopal R, Hewson R, et al. European perspective of 2-person rule for biosafety level 4 laboratories. Emerg Infect Dis. 2009;15:1858a–1860.
Lazzarini LCO, Spindola SM, Bang H, Gibson AL, Weisenberg S, Carvalho WDS, et al. RDRio mycobacterium tuberculosis infection is associated with a higher frequency of cavitary pulmonary disease. J Clin Microbiol. 2008;46:2175–83.
De Jong BC, Antonio M, Awine T, Ogungbemi K, De Jong YP, Gagneux S, et al. Use of spoligotyping and large sequence polymorphisms to study the population structure of the mycobacterium tuberculosis complex in a cohort study of consecutive smear-positive tuberculosis cases in the Gambia∇. J Clin Microbiol. 2009;47:994–1001.
Acknowledgments
The Research Support Foundation of Minas Gerais (FAPEMIG) (APQ 03266-13/APQ 00094-12) and the National Research Council (CNPq 446796/2014 and 310174/2017-7). The Postgraduate Program in Tropical Medicine and Infectology of the UFMG School of Medicine. The Ezequiel Dias Foundation (FUNED). The Oswaldo Cruz Foundation (Fiocruz) and the Brazilian Tuberculosis Research Network – REDE TB.
Funding
Funding was provided by The Research Support Foundation of Minas Gerais (FAPEMIG) (APQ 03266–13/APQ 00094–12) and the National Research Council (CNPq 446796/2014 and 310174/2017–7), as well as The Postgraduate Program in Tropical Medicine and Infectology of the UFMG School of Medicine and The Oswaldo Cruz Foundation (Fiocruz).
Author information
Authors and Affiliations
Contributions
SSM, PNS and WSC designed the study. INA, LJAF, NGTD, CJA performed the experiments. JPAH provided the statistical analysis. INA, SEGV and PNS were responsible genomic analysis. INA, SEGV and LJAF drafted the initial manuscript. PNS, WSC, SSM revised the manuscript, and all authors approved the final content of this manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the UFMG Research Ethics Committee (CAAE – 06611912.8.0000.5149). The consent to participate requirement was waived by the ethics committee, since clinical, sociodemographic and epidemiological data from patients were not included, and the studies were performed using DNA from culture and not extracted from clinical samples.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
The Genetic profile of Mycobacterium tuberculosis RDRio in Minas Gerais as defined by spoligotyping (DOCX 23 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
de Almeida, I.N., Vasconcellos, S.E.G., de Assis Figueredo, L.J. et al. Frequency of the Mycobacterium tuberculosis RDRio genotype and its association with multidrug-resistant tuberculosis. BMC Infect Dis 19, 556 (2019). https://doi.org/10.1186/s12879-019-4152-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-019-4152-7