
Abstract
Background
Although real-life results of sofosbuvir/simeprevir have been extensively reported from the United States, data from other geographical areas are limited. In the French observational cohort, ANRS CO22 HEPATHER, 9432 patients were given the new oral antivirals from December 2013 to June 30, 2018. We report the results of sofosbuvir/simeprevir in genotypes 1- and 4-infected patients.
Methods
Demographics and history of liver disease were collected at entry in the cohort. Clinical, adverse events, and virological data were collected throughout treatment and post-treatment follow-up. The choice of treatment duration or addition of ribavirin was left up to the physician.
Results
Five hundred ninety-nine HCV (467 genotype 1 and 132 genotype 4) mono-infected, naïve for all oral-DAAs regimen patients were given sofosbuvir/simeprevir with (n = 63) or without ribavirin (n = 536) for 12 or 24 weeks; 56% had cirrhosis (4% decompensated) and 71% had prior treatment failure to interferon-based regimen. 7 patients (1.16%) were lost to follow-up. The overall SVR12 rate was 92.6%. The SVR12 was 90% in GT1a, 94.2% in GT1b and 91.6% in GT4 with no significant difference for genotype, treatment duration or ribavirin addition. Severity of liver disease was not associated with a lower SVR12 rate on multivariate analysis but was associated with a higher rate of severe side effects. Early treatment discontinuations were rare; no new safety signals were reported.
Conclusion
In this real life, observational, prospective cohort study, the 12-week sofosbuvir/simeprevir+/−ribavirin combination appears to be efficient and safe.
Trial registration
Trial registration with ClinicalTrials.gov NCT01953458.
Similar content being viewed by others


Key points
-
The combination of sofosbuvir and simeprevir results in an overall SVR12 of 92.6, 90% in patients with genotype 1a infection, 94.2% with genotype 1b and 91.6% with genotype 4.
-
The safety of the sofosbuvir and simeprevir combination was acceptable with only 3% of early discontinuations and with no new safety warnings.
-
The combination with sofosbuvir and simeprevir is no longer recommended but remains a potential therapeutic option in resource-limited settings or in countries where simeprevir is still available.
Background
Chronic hepatitis C virus (HCV) infection is a worldwide disease that is responsible for hepatic and extrahepatic morbidity and mortality [1, 2]. A sustained virological response (SVR) corresponds to a complete cure of infection. A SVR is also associated with a reduction in HCV-related complications such as cirrhosis, hepatocellular carcinoma, the need for transplantation and death [3,4,5,6]. A better understanding of the viral cycle and characterization of the non-structural proteins of the virus led to development of direct acting antivirals (DAAs) against HCV [7,8,9]. Approved in the Spring of 2011, second generation DAAs replaced first generation protease inhibitors, which were then removed from the market in 2014. NS5B polymerase inhibitors (sofosbuvir, dasabuvir), protease inhibitors (simeprevir, paritaprevir, grazoprevir, glecaprevir, voxilaprevir) and NS5A replication complex inhibitors (daclatasvir, ledipasvir, ombitasvir, elbasvir, pibrentasvir and velpatasvir) have also been approved and evaluated [10,11,12,13,14,15,16,17,18,19,20,21]. A combination of pangenotypic drugs is now recommended to treat chronic HCV infection, while EASL guidelines no longer recommend the combination of sofosbuvir and simeprevir [22,23,24]. For the European Association for the Study of the Liver (EASL), antiviral therapy should be considered in all patients with chronic HCV infection because of the efficacy and safety profile of DAAs. Because of the long timelines for approval and in addition to clinical trials, preliminary real-life results of the combination of sofosbuvir/simeprevir have been extensively reported [25,26,27,28,29,30] from the United States (US) in patients with genotype 1 infection. However, real-life data from Europe or outside the US, and for genotype 4 are limited [31,32,33].
We report the real-life results of the French ANRS CO22 Hepather cohort for the sofosbuvir+simeprevir +/− ribavirin combination in patients with HCV genotypes 1 or 4 mono-infection.
Methods
Study design and participants
The ANRS CO22 HEPATHER cohort « Therapeutic option for hepatitis B and C: a French cohort » is a multicenter, national, prospective, observational cohort study of patients infected with hepatitis B or C virus (ClinicalTrials.gov registry number: NCT01953458). The cohort has been extensively described elsewhere [34].
In summary, by December 31, 2015, 20,798 patients had been included in the cohort, including 14,195 HCV-positive patients. A total of 9432 patients were given treatment including at least one direct acting antiviral from December 2013 to June 30, 2018. We selected all patients with HCV genotype 1 or genotype 4 infection who initiated a combination of sofosbuvir (400 mg/d) and simeprevir (150 mg/d) with or without ribavirin (1–1.2 g/d) before October 31, 2014 (n = 599). Patients who were liver transplant recipients, previously treated with other DAAs (except first generation protease inhibitors) or involved in clinical trials, were excluded. Patients were divided into four groups according to the scheduled duration of treatment and whether the regimen included ribavirin. This was an observational and not a randomized controlled study and the choice of treatment combination, duration and addition of ribavirin was left up to the physician. The diagnosis of cirrhosis was based either on the results of liver biopsy, a fibrotest result > 0.7 or fibroscan greater than 14.5 kPa. The duration of chronic hepatitis was estimated by the date of contamination, if available.
Outcomes
The main endpoint criterion was SVR at 12 weeks (SVR12) defined as undetectable HCV RNA 12 weeks after the last treatment. Secondary endpoints were undetectable HCV RNA 4 weeks after the last treatment (SVR4), early treatment discontinuation and adverse events.
Statistical analyses
A post-hoc calculation showed that the present study achieved a reliability of 2.4% for an anticipated 90% SVR12 and had a power > 80% for detecting Odds-Ratio (OR) < 0.3 for factors associated with SVR12, assuming the exposure to these factors ranged between 20 to 75%. Missing SVR12 measurements were determined using SVR24 measurements, if available (n = 52), otherwise using the SVR4 measurement (n = 6). Patients who died before SVR12 and therefore could not be evaluated (n = 4) and patients who were lost of follow-up (n = 7) and had no measurement of the virological response after the end of therapy, were considered to be virological failures.
Proportions were compared using the Fisher exact test and/or continuous outcomes were compared using the Kruskal-Wallis test. Comparisons with stratification for treatment duration or ribavirin use were tested using the Cochran-Mantel Haenszel (CMH) Chi-Square. Exact logistic regression models were used to assess independent baseline variables associated with SVR12 or serious side effects (including death). Predefined cut-offs were used to categorize all continuous factors. A univariate exact logistic model was estimated for each factor. Primary multivariate analysis included ribavirin (No vs Yes), treatment duration (12 vs. 24 weeks) and any factors with a P-value < 0∙10 on univariate analysis. A backward selection was applied retaining variables with a P-value < 0∙05. Statistical analyses were performed with SAS 9.4 software (SAS Institute Inc., Cary, North Carolina, USA).
Role of the funding source
The ANRS CO22 HEPATHER cohort is sponsored by Inserm-ANRS (French National Institute for Health and Medical Research – ANRS/France REcherche Nord&Sud Sida-hiv Hépatites). The sponsor contributed to the study design and writing of this report. The sponsor had no role in data collection, data analysis or data interpretation. The other sponsors of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. FC had full access to all the data in the study and SP and FC had the final responsibility for the decision to submit for publication.
Results
Patient population
By October 31, 2014, 599 cohort participants with HCV genotypes 1 (N = 467) or genotype 4 (N = 132) infection had started treatment with the sofosbuvir/simeprevir combination including 536 (89%) who did not receive and 63 (11%) who did receive ribavirin based on the physician’s decision (Fig. 1). The duration of treatment was 12 weeks in 530 (530/599 = 88.4%) patients and 24 weeks in 69 (69/599 = 11.5%).

Flow diagram
Patient characteristics are described in Table 1. Fifty-four percent were men, mean age 61 years old, 56% with cirrhosis, 4% with decompensated cirrhosis and 71% with interferon-based treatment failure (all were naïve for all oral DAAs regimens). Patients were infected with genotype 1a (23.5%), 1b (49.5%), 1 but unclassified (5%) or 4 (22%). The mean MELD score in the 312 with available data out of 335 patients with cirrhosis was 9.5 +/− 4.3 and 91.6% were Child-Pugh score A. Patients who received the 12-week combination of sofosbuvir/simeprevir differed from those who received the 24-week or ribavirin-containing regimens, with lower rates of cirrhosis (54% (259/478) vs 67% (76/114), p = 0.0158), decompensated cirrhosis (3% (16/485) vs 7% (8/114), p = 0.1051) and biochemical markers of liver severity: albumin< 30 g/L in 3% (12/437) vs 7% (7/101), p = 0.0650, prothrombin time ≤ 70% in 10% (45/449) vs 21% (23/110), p = 0.0031, thrombocyte count < 100,000/mm3 in 19% (87/468) vs 31% (34/111), p = 0.064 and conjugated bilirubinemia ≥5 μmol/L in 52% (133/257) vs 67% (49/73), p = 0.0233.
Efficacy
Missing SVR12 measurements were determined using SVR24 measurements in 52 patients and SVR4 measurements in 6 patients. Four patients who died before reaching SVR12 and seven patients who were lost to follow-up with no virological response measurement at the end of therapy were considered virological failures.
A SVR12 was achieved in a total of 555 (92.6%) patients. SVR12 rates ranged from 89% in patients who received a 24-week sofosbuvir/simeprevir/ribavirin combination regimen to 98% in patients who received a 12-week sofosbuvir/simeprevir/ribavirin combination regimen (Table 2 and Fig. 2).

Percentages of SVR12 in subgroups of patients by genotype
Although no significant difference was found between the 12- or 24-week regimen with or without ribavirin, the groups were not comparable because patients in the ribavirin and 24-week treatment groups had more severe disease.
The SVR12 rate in patients without cirrhosis was 94.9%, whatever the treatment.
The SVR12 rate in patients with cirrhosis was 90.7% in treatment-naïve and 89% in treatment-experienced patients. No significant difference was found between those who received a 24-week regimen and a 12-week regimen (42/49 (85.7% %) versus 262/286 (91.6%), respectively (CMH Chi Square stratified on ribavirin containing regimen: p = 0.12)). The SVR12 rate was not different between those who received a 12-week regimen with or without ribavirin (26/27 (96%) versus 236/259 (91%), respectively (CMH Chi Square stratified on ribavirin containing regimen: p = 0.39)).
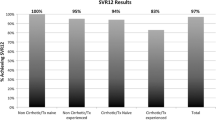
The overall SVR12 was 90, 94.2 and 91.6% in patients with genotypes 1a, 1b and 4, respectively, and was not significant different among the groups (Table 3). The addition of ribavirin or not, and the duration of treatment (12 vs 24 weeks) did not influence the SVR12 rate.
There were no variables associated with SVR12 at the 0.05 level on univariate analysis (Table 3).
Age, sex, gender, BMI, genotype, prior treatment history, cirrhosis or not and treatment duration did not influence SVR12. No factor was associated with the absence of SVR12 on multivariate analysis.
Safety and tolerability
Early treatment discontinuation only occurred in 18 patients (3%). The rate of discontinuation for adverse events was 1.5%. The rate of discontinuation was higher in patients treated for 24 weeks or with ribavirin (Table 4). Nine of the patients who discontinued treatment (50%) achieved a SVR12. Eight (44%) had been treated for 8 weeks or more.
Four patients died during follow-up. One patient died from a subdural hematoma in the first week after initiating treatment, and one patient at week 8 from undetermined causes. Two patients died suddenly at week 12 from cardiac arrest, which was considered to be possibly treatment-related (sofosbuvir/simeprevir). Cardiovascular side effects (mainly bradyarrhytmias) have been associated with sofosbuvir treatment and may result in the implementation of a pace-maker [35] and associated with a risk of sudden, unexplained death [36].
Forty-three other serious adverse events occurred in 37 (9%) patients with no difference between treatment with or without ribavirin, but with a higher rate in the 24- week regimen (p = 0.0453). Two of these serious adverse events were considered to be possibly treatment-related (simeprevir): one malaise at week 12 and one drug-induced acute hepatitis at week 5.
The most common adverse events (≥10% in any subgroup) were asthenia, headache and pruritus.
Univariate analysis identified treatment duration, prothrombin time ≤ 70%, decompensated cirrhosis, time since diagnosis ≥15 years, MELD ≥15 or cirrhosis at inclusion, cirrhosis, platelet count < 100,000/mm3, conjugated bilirubin ≥5 μmol/L and albumin < 30 g/L as potential predictors of serious adverse events. A prothrombin time ≤ 70% (OR versus prothrombin time ≥ 70%, 2.88 95%CI 1.24–6.48; P = 0.0127), MELD ≥15 or cirrhosis (OR versus MELD < 15 and no cirrhosis, 3.13 95%CI 1.2–9.62; P = 0.0154) and a time since diagnosis ≥15 years (OR versus time since diagnosis < 15 years, 2.19 95%CI 1.01–5.1; P = 0.0465) remained the only 3 factors independently associated with serious adverse events. It should be noted that age and gender were not associated with serious adverse events.
Discussion
Although the real-life results of the sofosbuvir/simeprevir combination have been extensively reported in US genotype 1-infected patients, data from other geographical areas or other genotypes are limited. In this real-life study, we analyzed the efficacy and safety of the sofosbuvir+simeprevir +/− ribavirin combination in patients with genotypes 1 or 4 infection from the French ANRS CO22 HEPATHER cohort, a real-life study. Most of these patients were “difficult-to-treat” since 56% had cirrhosis, 4% had decompensated cirrhosis, 71% had failed prior treatment with pegylated interferon and ribavirin and 5% associated with telaprevir or boceprevir. Only 7 patients (1.16%) were lost to follow-up with no available PCR after the end of treatment. The sofosbuvir/simeprevir combination resulted in a global SVR12 rate of 92.6%. Most patients (81%) received the sofosbuvir/simeprevir combination without ribavirin for 12 weeks with a SVR of 93%. No significant difference in SVR12 rate was found between 12 or 24 weeks of treatment, with or without ribavirin. However, it is not possible to conclude whether extending the duration of treatment or the addition of ribavirin is needed or not, especially in patients with cirrhosis or decompensated cirrhosis because of the small sample size and because patients had more severe liver disease in the ribavirin and 24- week regimen groups. The overall SVR12 was 94.2% in patients with genotype 1b infection (vs 90 and 91.6% for genotypes 1a and 4, respectively) but the difference was not statistically significant. There was no factor associated with treatment failure on univariate analysis.
Early treatment discontinuation was rare and no new safety signals were reported compared to previous studies. The severity of liver disease (MELD ≥15 or cirrhosis at inclusion) was a risk factor for serious adverse events which support a causal relationship between adverse events and protease inhibitor exposure, as previously reported in the CUPIC study [37].
It is not possible to compare our results with those of other studies, clinical trials or real-world studies, because the rate of “difficult to treat” patients differed. The SVR12 rate in the real-world US TARGET cohort [25] including 59% of patients with cirrhosis (56% in our study) and around 50% of treatment-experienced patients (71% in our study) was 84%. In that study, in contrast to our results, the severity of liver disease and previous protease inhibitor treatments were associated with treatment failure in the model-adjusted estimates. In an Egyptian [32] and a US real-life study [38] the SVR rate was > 92%. In summary, our study shows a SVR-rate of nearly 95% in patients without cirrhosis, and 91% in those with cirrhosis, which is comparable to other real-life studies [31,32,33].
Even with a SVR rate of 91–95%, the role of simeprevir/sofosbuvir, is debatable with the current high turnover of DAAs. In the era of “second” wave DAAs (sofosbuvir with ledipasvir, daclatasvir or velpatasvir and voxilaprevir, ombitasvir/paritaprevir/dasabuvir, grazoprevir/elbasvir, glecaprevir/pibrentasvir) with high SVR rates in both clinical trials and real-life studies [34] and access to shorter-duration pangenotypic regimens even in patients with cirrhosis, our real-world study suggests that the efficacy of the 12-week sofosbuvir/simeprevir combination is probably suboptimal despite an acceptable safety profile.
Conclusion
This study reports the real-life results of the French ANRS CO22 Hepather cohort for the sofosbuvir+simeprevir +/− ribavirin combination in patients with HCV genotypes 1 or 4 mono-infection. The overall SVR12 was 92.6, 90% in patients with genotype 1a infection, 94.2% with genotype 1b and 91.6% with genotype 4 with an acceptable safety profile. In the era of “second” wave DAAs this combination is no longer recommended in the most recent (2018) EASL guidelines but could remain a therapeutic option in low-income countries without access to pangenotypic drugs.
Abbreviations
- BMI:
-
Body mass index
- DAA:
-
Direct-acting antiviral
- EASL:
-
European Association for the Study of the Liver
- HCV:
-
Hepatitis C virus
- PCR:
-
Polymerase chain reaction
- PEG:
-
Pegylated interferon
- PI:
-
Protease inhibitor
- RBV:
-
Ribavirin
- RNA:
-
Ribonucleic acid
- SD:
-
Standard deviation
- SMV:
-
Simeprevir
- SOF:
-
Sofosbuvir
- SVR:
-
Sustained virological response
References
Younossi ZM, Kanwal F, Saab S, Brown KA, El-Serag HB, Kim WR, et al. The impact of hepatitis C burden: an evidence-based approach. Aliment Pharmacol Ther. 2014;39(5):518–31.
Marcellin P, Pequignot F, Delarocque-Astagneau E, Zarski JP, Ganne N, Hillon P, et al. Mortality related to chronic hepatitis B and chronic hepatitis C in France: evidence for the role of HIV coinfection and alcohol consumption. J Hepatol. 2008;48(2):200–7.
Deuffic-Burban S, Deltenre P, Louvet A, Canva V, Dharancy S, Hollebecque A, et al. Impact of viral eradication on mortality related to hepatitis C: a modeling approach in France. J Hepatol. 2008;49(2):175–83.
Backus LI, Boothroyd DB, Phillips BR, Belperio P, Halloran J, Mole LA. A sustained virologic response reduces risk of all-cause mortality in patients with hepatitis C. Clin Gastroenterol Hepatol. 2011;9(6):509–16.e1.
van der Meer AJ, Veldt BJ, Feld JJ, Wedemeyer H, Dufour JF, Lammert F, et al. Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis. Jama. 2012;308(24):2584–93.
Nahon P, Bourcier V, Layese R, Audureau E, Cagnot C, Marcellin P, et al. Eradication of hepatitis C virus infection in patients with cirrhosis reduces risk of liver and non-liver complications. Gastroenterology. 2017;152(1):142–56.e2.
Sofia MJ, Bao D, Chang W, Du J, Nagarathnam D, Rachakonda S, et al. Discovery of a beta-d-2′-deoxy-2′-alpha-fluoro-2′-beta-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem. 2010;53(19):7202–18.
Raboisson P, de Kock H, Rosenquist A, Nilsson M, Salvador-Oden L, Lin TI, et al. Structure-activity relationship study on a novel series of cyclopentane-containing macrocyclic inhibitors of the hepatitis C virus NS3/4A protease leading to the discovery of TMC435350. Bioorg Med Chem Lett. 2008;18(17):4853–8.
Penin F, Dubuisson J, Rey FA, Moradpour D, Pawlotsky JM. Structural biology of hepatitis C virus. Hepatology (Baltimore, Md). 2004;39(1):5–19.
Lawitz E, Mangia A, Wyles D, Rodriguez-Torres M, Hassanein T, Gordon SC, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med. 2013;368(20):1878–87.
Lawitz E, Sulkowski MS, Ghalib R, Rodriguez-Torres M, Younossi ZM, Corregidor A, et al. Simeprevir plus sofosbuvir, with or without ribavirin, to treat chronic infection with hepatitis C virus genotype 1 in non-responders to pegylated interferon and ribavirin and treatment-naive patients: the COSMOS randomised study. Lancet (London, England). 2014;384(9956):1756–65.
Gane EJ, Stedman CA, Hyland RH, Ding X, Svarovskaia E, Symonds WT, et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med. 2013;368(1):34–44.
Sulkowski MS, Gardiner DF, Rodriguez-Torres M, Reddy KR, Hassanein T, Jacobson I, et al. Daclatasvir plus sofosbuvir for previously treated or untreated chronic HCV infection. N Engl J Med. 2014;370(3):211–21.
Kowdley KV, Gordon SC, Reddy KR, Rossaro L, Bernstein DE, Lawitz E, et al. Ledipasvir and sofosbuvir for 8 or 12 weeks for chronic HCV without cirrhosis. N Engl J Med. 2014;370(20):1879–88.
Kowdley KV, Lawitz E, Poordad F, Cohen DE, Nelson DR, Zeuzem S, et al. Phase 2b trial of interferon-free therapy for hepatitis C virus genotype 1. N Engl J Med. 2014;370(3):222–32.
Zeuzem S, Jacobson IM, Baykal T, Marinho RT, Poordad F, Bourliere M, et al. Retreatment of HCV with ABT-450/r-ombitasvir and dasabuvir with ribavirin. N Engl J Med. 2014;370(17):1604–14.
Kwo P, Gitlin N, Nahass R, Bernstein D, Etzkorn K, Rojter S, et al. Simeprevir plus Sofosbuvir (12 and 8 weeks) in HCV genotype 1-infected patients without cirrhosis: OPTIMIST-1, a phase 3, randomized study. Hepatology (Baltimore, Md). 2016;64(2):370–80.
Lawitz E, Matusow G, DeJesus E, Yoshida EM, Felizarta F, Ghalib R, et al. Simeprevir plus sofosbuvir in patients with chronic hepatitis C virus genotype 1 infection and cirrhosis: a phase 3 study (OPTIMIST-2). Hepatology (Baltimore, Md). 2015;64(2):360–69.
Kwo PY, Poordad F, Asatryan A, Wang S, Wyles DL, Hassanein T, et al. Glecaprevir and pibrentasvir yield high response rates in patients with HCV genotype 1-6 without cirrhosis. J Hepatol. 2017;67(2):263–71.
Feld JJ, Jacobson IM, Hezode C, Asselah T, Ruane PJ, Gruener N, et al. Sofosbuvir and Velpatasvir for HCV genotype 1, 2, 4, 5, and 6 infection. N Engl J Med. 2015;373(27):2599–607.
Bourliere M, Gordon SC, Flamm SL, Cooper CL, Ramji A, Tong M, et al. Sofosbuvir, Velpatasvir, and Voxilaprevir for previously treated HCV infection. N Engl J Med. 2017;376(22):2134–46.
EASL. Recommendations on treatment of hepatitis C 2015. J Hepatol. 2015;63(1):199–236.
AASLD. HCV guidance : Recommendations for testing, managing, and treating hepatitis C. 2015.
EASL. Recommendations on treatment of hepatitis C 2018. J Hepatol. 2018;69(2):461–511.
Sulkowski MS, Vargas HE, Di Bisceglie AM, Kuo A, Reddy KR, Lim JK, et al. Effectiveness of Simeprevir plus Sofosbuvir, with or without ribavirin, in real-world patients with HCV genotype 1 infection. Gastroenterology. 2016;150(2):419–29.
Shiffman ML, James AM, Long AG, Alexander PC. Treatment of chronic HCV with sofosbuvir and simeprevir in patients with cirrhosis and contraindications to interferon and/or ribavirin. Am J Gastroenterol. 2015;110(8):1179–85.
Saxena V, Nyberg L, Pauly M, Dasgupta A, Nyberg A, Piasecki B, et al. Safety and efficacy of Simeprevir/Sofosbuvir in hepatitis C-infected patients with compensated and decompensated cirrhosis. Hepatology (Baltimore, Md). 2015;62(3):715–25.
Backus LI, Belperio PS, Shahoumian TA, Loomis TP, Mole LA. Effectiveness of sofosbuvir-based regimens in genotype 1 and 2 hepatitis C virus infection in 4026 U.S. veterans. Aliment Pharmacol Ther. 2015;42(5):559–73.
Aqel BA, Pungpapong S, Leise M, Werner KT, Chervenak AE, Watt KD, et al. Multicenter experience using simeprevir and sofosbuvir with or without ribavirin to treat hepatitis C genotype 1 in patients with cirrhosis. Hepatology (Baltimore, Md). 2015;62(4):1004–12.
Dieterich D, Bacon BR, Flamm SL. Evaluation of sofosbuvir and simeprevir-based regimens in the trio network: academic and community of a real-world, heterogenous population. Hepatology (Baltimore, Md). 2014;60(Suppl 1):220A–220A.
Willemse SB, Baak LC, Kuiken SD, van der Sluys VA, Lettinga KD, van der Meer JT, et al. Sofosbuvir plus simeprevir for the treatment of HCV genotype 4 patients with advanced fibrosis or compensated cirrhosis is highly efficacious in real life. J Viral Hepat. 2016;1–5.
El-Khayat HR, Fouad YM, Maher M, El-Amin H, Muhammed H. Efficacy and safety of sofosbuvir plus simeprevir therapy in Egyptian patients with chronic hepatitis C: a real-world experience. Gut. 2017;66(11):2008–12.
Marino Z, Pascasio-Acevedo JM, Gallego A, Diago M, Baliellas C, Morillas R, et al. High efficacy of Sofosbuvir plus Simeprevir in a large cohort of Spanish cirrhotic patients infected with genotypes 1 and 4. Liver Int. 2017;37(12):1823–32.
Pol S, Bourliere M, Lucier S, Hezode C, Dorival C, Larrey D, et al. Safety and efficacy of daclatasvir-sofosbuvir in HCV genotype 1-mono-infected patients. J Hepatol. 2017;66(1):39–47.
Fontaine H, Lazarus A, Pol S, Pecriaux C, Bagate F, Sultanik P, et al. Bradyarrhythmias associated with Sofosbuvir treatment. N Engl J Med. 2015;373(19):1886–8.
Laurain A, Kramer L, Sultanik P, Vallet-Pichard A, Sogni P, Pol S. Mortality associated with the treatment of HCV with direct-acting antivirals. Gut. 2018;67(1):197–8.
Hezode C, Fontaine H, Dorival C, Larrey D, Zoulim F, Canva V, et al. Triple therapy in treatment-experienced patients with HCV-cirrhosis in a multicentre cohort of the French early access Programme (ANRS CO20-CUPIC) - NCT01514890. J Hepatol. 2013;59(3):434–41.
Alam I, Brown K, Donovan C, Forlenza J, Lauwers K, Mah'moud MA, et al. Real-world effectiveness of Simeprevir-containing regimens among patients with chronic hepatitis C virus: the SONET study. Open Forum Infect Dis. 2017;4(1):ofw258.
Acknowledgments
Study investigators Delphine Bonnet, Virginie Sicart (CHU Purpan, Toulouse, France), François Bailly, Marjolaine Beaudoin, Dominique Giboz, Kerstin Hartig-Lavie, Marianne Maynard (Hospices Civils de Lyon, Lyon, France), Morane Cavellec, Marjorie CheraudCarpentier, François Raffi, Florian Vivrel, Eric Billaud (Hôpital Hôtel-Dieu, France), Jaouad Benhida, Jérôme Boursier, Paul Calès, Françoise Lunel, Frédéric Oberti, Isabelle Hubert-Fouchard, (CHU Angers, Angers, France), Nathalie Boyer, Audrey Gilibert, Nathalie Giuily (Hôpital Beaujon, Clichy, France), Giovanna Scoazec (Hôpital Beaujon, Clichy, France and Hôpital Henri Mondor, Créteil, France), Sandrine Fernandes, Sylvie Keser, Philippe Sultanik, Anaïs Vallet-Pichard (Hôpital Cochin, Paris, France), Juliette Foucher, Jean-Baptiste Hiriart, Aurore Mathias, Julien Vergniol (Hôpital HautLévêque, Pessac, Bordeaux, France), Chrystelle Ansaldi, Laëtitia Chouquet, Emilie De Luca, Valérie Oules (Hôpital Saint Joseph, Marseille, France), Rodolphe Anty, Eve Gelsi, Régine Truchi (CHU de Nice, Nice, France), Elena Luckina, Nadia Messaoudi, Joseph Moussali, (Groupe Hospitalier Pitié-Salpétrière, Paris, France), Barbara De Dieuleveult, Damien Labarriere, Pascal Potier, Si Nafa Si Ahmed, Xavier Causse (CHR La Source, Orléans, France), Nathalie Ganne-Carrié, Véronique Grando-Lemaire, Pierre Nahon, Alan Peltier, Judith Ung, Valérie Bourcier (Hôpital Jean Verdier, Bondy, France), Mariette Gougeon, Anne Guillygomarch, Caroline Jezequel (CHU Rennes, Rennes, France), Romain Moirand, Thomas F. Baumert, Michel Dofföel, Catherine Mutter, Pauline Simo-Noumbissie, François Habersetzer (Hôpitaux Universitaires de Strasbourg, Strasbourg, France), Hélène Barraud, Mouni Bensenane, Abdelbasset Nani, Sarah Hassani-Nani (CHU de Nancy, Vandoeuvre-lès-Nancy, France), Marie-Albertine Bernard (CHU de Nancy, Vandoeuvre-lès-Nancy, France and Centre Hospitalier Régional, Metz, France), Michael Bismuth, Ludovic Caillo, Stéphanie Faure, Georges-Philippe 3 Pageaux, Marie Pierre Ripault (Hôpital Saint Eloi, Montpellier, France), Karl Barange, Christophe Bureau, Jean Marie Peron, Marie Angèle Robic, Léa Tarallo (CHU Purpan, Toulouse, France), Marine Faure, Bruno Froissart, Marie-Noelle Hilleret, Vincent Leroy (CHU de Grenoble, Grenoble, France), Odile Goria, Victorien Grard, Hélène Montialoux (CHU Charles Nicolle, Rouen, France), Muriel François, Christian Ouedraogo, Christelle Pauleau, Anne Varault (Hôpital Henri Mondor, Créteil, France), Tony Andreani, Bénédicte Angoulevant, Azeline Chevance, Lawrence Serfaty (Hôpital Saint-Antoine, Paris, France), Teresa Antonini, Audrey Coilly, Jean-Charles Duclos Vallée, Mariagrazia Tateo (Hôpital Paul Brousse, Villejuif, France), Armand Abergel, Corinne Bonny, Chanteranne Brigitte, Géraldine Lamblin, Léon Muti (Hôpital Estaing, Clermont-Ferrand, France), Abdenour Babouri, Virginie Filipe (Centre Hospitalier Régional, Metz, France), Camille Barrault, Laurent Costes, Hervé Hagège, Soraya Merbah (Centre Hospitalier Intercommunal, Créteil, France), Paul Carrier, Maryline Debette-Gratien, Jérémie Jacques (CHU Limoges, Limoges, France), Florent Artu, Valérie Canva, Sébastien Dharancy, Alexandre Louvet (CHRU Claude Huriez, Lille, France), Marc Bardou, Donya Da Costa Souhiel, Patrick Hillon, Marianne Latournerie (Dijon University Hospital, Dijon, France), Yannick Bacq, Didier Barbereau, Charlotte Nicolas (CHU Trousseau, 37044 Tours, France), Nisserine Ben Amara, Danièle Botta-Fridlund, Isabelle Portal (CHU Timone, Marseille, France), Moana Gelu-Simeon, Marie-Josée Lafrance (CHU de Pointe-à-Pitre, Pointe-à-Pitre, Guadeloupe).
Funding
ANRS (France Recherche Nord&sud Sida-vih Hepatites), ANR (Agence Nationale de la Recherche), DGS (Direction Générale de la Santé) and MSD, Janssen, Gilead, Abbvie, BMS, Roche.The biobank of the cohort is stored by Cell&Co Biorepository, Pont du Château, France and has been managed temporarily by Centre de Ressources Biologiques-HUEP, Hôpital St Antoine, Paris, France.
Availability of data and materials
The data that support the findings of this study are available from ANRS-INSERM but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. However, data are available from the authors upon reasonable request and with permission of ANRS-INSERM.
Author information
Authors and Affiliations
Consortia
Contributions
Study concept and design: FC, CD, HF, VPZ, SP. Acquisition of data: HF, CD, SM, GH, DL, CD, CH, FZ, PMar, MB, JPZ, DT, LA, NGC, PC, JPB, GR, CG, XC, AA, OC, PMat, DG, DS, AT, VLR, VPZ, AD, FC, SP. Analysis and interpretation of data: AL, GH, HF, FC, SP. Drafting of the manuscript: AL, SP. Critical revision of the manuscript for important intellectual content: All authors. Statistical analysis: GH, FC, CLN. Obtained funding: FC, HF, CD, VPZ, SP. Administrative, technical, or material support: FC, HF, CD, VPZ, SP. Study supervision: FC, HF, CD, VPZ, SP. Approval of the final version of the manuscript: all.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from each patient before enrolment. The protocol was performed in accordance with the Declaration of Helsinki and French law for biomedical research and was approved by the “CPP Ile de France 3” Ethics Committee (Paris, France) and the French Regulatory Authority (ANSM).
Consent for publication
not applicable.
Competing interests
The ANRS CO22 HEPATHER cohort is sponsored and funded by Inserm-ANRS and conducted in collaboration with Association Française pour l’étude du Foie (AFEF). The cohort received supports from ANR (Agence Nationale de la Recherche), DGS (Direction Générale de la Santé) and MSD, Janssen, Gilead, Abbvie, BMS, Roche. The public/private partnership is built in total transparency through a specific contract. The pharmaceutical companies are not involved in the scientific decisions.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Laurain, A., Metivier, S., Haour, G. et al. Safety and efficacy of the combination simeprevir-sofosbuvir in HCV genotype 1- and 4-mono-infected patients from the French ANRS CO22 hepather cohort. BMC Infect Dis 19, 300 (2019). https://doi.org/10.1186/s12879-019-3923-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-019-3923-5