
Abstract
Background
Genetic mutations that reduce intracellular superoxide production by granulocytes causes chronic granulomatous disease (CGD). These patients suffer from frequent and severe bacterial and fungal infections throughout their early life. Diagnosis is usually made in the first 2 years of life but is sometimes only diagnosed when the patient is an adult although they may have suffered from symptoms since childhood.
Case presentation
A 26-year-old man was referred with weight loss, fever, hepatosplenomegaly and coughing. He had previously been diagnosed with lymphadenopathy in the neck at age 8 and prescribed anti-tuberculosis treatment. A chest radiograph revealed extensive right-sided consolidation along with smaller foci of consolidation in the left lung. On admission to hospital he had respiratory problems with fever. Laboratory investigations including dihydrorhodamine-123 (DHR) tests and mutational analysis indicated CGD. Stimulation of his isolated peripheral blood neutrophils (PMN) with phorbol 12-myristate 13-acetate (PMA) produced low, subnormal levels of reactive oxygen species (ROS). Aspergillus terreus was isolated from bronchoalveolar lavage (BAL) fluid and sequenced.
Conclusions
We describe, for the first time, the presence of pulmonary A. terreus infection in an adult autosomal CGD patient on long-term corticosteroid treatment. The combination of the molecular characterization of the inherited CGD and the sequencing of fungal DNA has allowed the identification of the disease-causing agent and the optimal treatment to be given as a consequence.
Similar content being viewed by others

Background
Chronic granulomatous disease (CGD) is a rare inherited primary immune deficiency disorder [1,2,3]. It presents as life-threatening recurrent fungal and bacterial infections of the skin, lungs and bones with associated chronic inflammation or granulomas. As its name suggests, granulocytes (neutrophils and eosinophils) along with monocytes and macrophages are the major cellular targets. These cells have reduced phagocysis due to defective generation of reactive oxygen species (ROS) which are required to kill selective types of fungi and bacteria [1,2,3]. CGD is generally diagnosed in infancy or early childhood [4, 5].
Genetic defects in the nicotinamide adenine dinucleotide phosphate (NAPDH) oxidase complex can cause CGD. The 5 structural components that are mutated are the 3 cytoplasmatic components p47phox, p67phox and p40phox and the 2 membrane-associated glycoproteins gp91phox (phagocyte oxidase) and p22phox [6]. As a result, cells cannot eliminate infectious agents due to an inability to produce high levels of ROS. Subsequently, infections such as pneumonia, lymphadenitis, cutaneous and hepatic abscesses, osteomyelitis and septicemia are frequently observed and generally severe in nature [7]. The most commonly found infectious agents include Aspergillus species, Burkholderia cepacia complex, Candida, enteric gram-negative bacteria, Mycobacterium tuberculosis, marcescens and Staphylococcus aureus [8]. In addition, Aspergillus species such as A. fumigatus, A. flavus and A. nidulans are commonly isolated from CGD patients with fungal spondylodiscitis [9].
We describe here a case of autosomal CGD diagnosed in a 28-year-old male. He initially presented with granulomatous disease mimicking sarcoidosis and was treated with corticosteroids for 12 years. On admission to hospital he had respiratory problems with fever, and laboratory investigations including dihydrorhodamine-123 (DHR) tests [10] and mutational analysis indicated CGD. Aspergillus terreus was isolated from bronchoalveolar lavage (BAL) fluid and its presence was confirmed by sequencing.
Case presentation
A 26-year-old man with a history of consanguineous parents (cousins) was referred with weight loss, fever, hepatosplenomegaly and coughing. He had previously been diagnosed with lymphadenopathy in the neck at age 8 and prescribed anti-tuberculosis treatment. At 12-years of age he was diagnosed with pulmonary sarcoidosis and corticosteroid treatment was initiated.
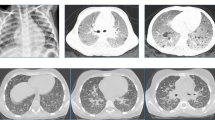
On examination on the day of admission to our hospital the patient was pale with low-grade pyrexia (37.5 °C). Cardiovascular examinations were normal but he had cervical lymphadenopathy. A chest radiograph revealed extensive right-sided consolidation along with smaller foci of consolidation in left lung (Fig. 1a). Crackles were heard on the left side.

Lung imaging. a. There is extensive consolidation in the right lung (red arrow). Scattered patches of consolidation in the left lung are also seen (green arrow). Chest CT scans show the lung window at the level of the pulmonary artery (b) and heart (c). Note extensive consolidation in the right upper lobe (red arrow b). There are also smaller patches of consolidation in the right lower and middle lobes and in the left lung (green arrows b and c)
Moxifloxacin treatment for 2 weeks did not alleviate his cough, hypoxia or night sweats and fevers of up to 40 °C. Thoracic computed tomography demonstrated mediastinal lymphadenopathy and bilateral consolidation that was greater in the right lung. Non-specific inflammation was shown in a lung biopsy (Fig. 1b and c). Full blood counts were normal and liver function tests and autoimmune and virology screens were negative.
Whole blood was examined with specific laboratory tests for neutrophil NADPH oxidase activity i.e. nitroblue tetrazolium (NBT) [10] and DHR tests [10,11,12,13,14]. Low, subnormal levels of ROS were produced following stimulation of purified peripheral blood neutrophils (PMN) with phorbol 12-myristate 13-acetate (PMA) (Fig. 2).

Dihydrorhodamine-1,2,3 (DHR) analysis of reactive oxygen species generation by peripheral blood neutrophils. Patient and healthy control cells were incubated with DHR (375 ng/ml), with or without PMA (100 ng/ml), and ROS generation was assayed by FACS analysis. The mean fluorescent intensity (MFI) of the following groups are indicated in the figure: Red lines represent cells from unstimulated healthy controls and patient cells, blue line represents PMA-stimulated patient cells, and grey line represents and PMA-stimulated healthy control cells
BAL and serum galactomannan (GAM) tests were negative but the BAL sample was sent for microbiological analysis. 48-72 h culture of the BAL sample on sabouraud dextrose agar at 30 °C resulted in the appearance of smooth light yellow powdery colonies that became darker over time (Fig. 3a). Lactophenol cotton blue (LPCB) mounting medium slide culture was performed for microscopic species identification (Fig. 3b and c). Microscopic analysis revealed septate and hyaline hyphae with biseriate phialides extending from the upper portion of the vesicle and covering 2/3 of the plate. Hyaline, globose or oval and thick-walled chlamydoconidia were also seen (Fig. 3b and c). The isolates were identified by phenotypic (macroscopic and microscopic) characteristics as Aspergillus terreus, and this identification was confirmed by DNA sequencing.

Microbiological culture of lung fungal species obtained by bronchoscopy. a 7-day-old culture on sabouraud dextrose agar at 30 °C shows light yellow to brownish colonies. b Fungal growth after 12 days sterile culture on potato dextrose agar (PDA) plates for sporulation and identification, and LPCB mounting slide showing details of hyphae and the accessory conidia. c Septate and hyaline hyphae with biseriate phialides extending from the upper portion of the vesicle and covering 2/3 of the plate
The fungal culture was disrupted with glass beads in a grinder and DNA was extracted with phenol chloroform. DNA was suspended in 50 μl of double distilled water and stored at − 20 °C for future use [1, 2]. The beta tubulin gene was amplified with forward (Bta2a: 5′-GGTAACCAAATCGGTGCTGCTTTC-3′) and reverse (Bta2b 5′-ACCCTCAGTGTAGTGACCCTTGGC-3′) primers and sequenced. The DNA sequence results were compared against the NCBI Genebank database, which showed a 99% similarity with an Aspergillus terreus isolate in the Gene Bank fungal library with accession no 1168 [15, 16] (Fig. 4).

a Partial sequence of the putative invertase gene from Aspergillus terreus isolated from the patient. This is compared to the sequence of the ‘standard’ A. terreus sp. (b)
Genomic DNA was extracted from the blood mononuclear cell fraction using a Gentra Puregene Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. DNA sequencing was performed at the Sanquin Research Laboratory (Amsterdam, The Netherlands). GeneScan was used to determine the ratio between the number of exon 2 sequences of neutrophil cytosolic factor 1 (NCF1) gene, which encodes p47phox, and the number of Ψ-NCF1 exon 2 sequences [17]. This revealed a homozygous GT deletion (c.75_76delGT) at the start of exon 2 in NCF1, resulting in the introduction of a frame shift and a premature stop codon (p.Tyr26HisfsTer26) predicting a truncated and inactive p47phox protein.
The patient was treated with meropenem, vancomycin and liposomal amphotericin b for 2 weeks. After obtaining the results of fungal characterization, voriconazole was started and other antibiotics were removed from the treatment strategy. After 3 months, the patient had recovered as confirmed by chest imaging and clinical manifestations. The patient gained 10 kg in weight and is on maintenance treatment with voriconazole.
Discussion and conclussions
We report here an adult autosomal CGD subject with pulmonary A. terreus infection. We are unaware of any previous cases describing pulmonary A. terreus infection in an adult on long-term corticosteroid treatment. We combined molecular identification of CGD mutation with fungal DNA sequencing to correctly identify the causal agent and consequently provided optimal therapy for the patient. In the first 3 months of treatment the patient gained 10 kg in weight indicating the success of the diagnosis and treatment.
Most pulmonary fungal infections in CGD patients are due to A. fumigatus although A. terreus can cause pneumonia and disseminated infections in man [18,19,20,21,22,23,24,25]. In humans, A. fumigatus, A. flavus and A. niger are much more common pathogens than A. terreus [25] although A. terreus has been associated with vertebral infections [26,27,28,29]. Immunocompromised individuals are at most risk of opportunistic infection by A. terreus. These patients include COPD patients on corticosteroids, cancer patients receiving chemotherapy and patients with HIV/AIDS [29,30,31].
It is likely, therefore that the A. terreus infection seen in this CGD patient with an inherited defect of PMN function and on long-term treatment with corticosteroids was opportunistic. The incidence of A. terreus has risen from < 2% in 1996 to 15% in 2001 [32]. The resistance of A. terreus to amphotericin B, thermotolerance and the production of accessory conidia have been proposed as mechanisms that explain the rapid dissemination of the organism during invasive infections [33].
A. Terreus is an opportunistic fungus with an increasing prevalence amongst aspergillosis species and has a higher mortality rate (51% versus 30%) compared to other species [34,35,36,37]. A. Terreus infection often leads to superficial infections in man whereby the inhalation of spores initiates the immediate recruitment and activation of macrophages and neutrophils that overwhelms the infection [38]. However, in immunosuppressed individuals with neutropenia, or with a compromised capacity for neutrophil phagocytosis, this response is less vigorous [39].
Most CGD patients are susceptible to opportunistic infections. Indeed, the most important symptom of this disease is recurrent and/or invasive infections, especially those caused by catalase-positive bacteria and fungi. In Western countries, about 30% of CGD cases have been reported to be autosomal recessive (AR), of which the majority is due to mutations in NCF1 [5, 40]. However, in a recent multicenter study conducted in Iran, the majority of CGD patients were found to be AR (87%) and only a minority had X-linked CGD (13%) [41]. It is important in regions where tuberculosis is endemic, such as Iran, that sarcoidosis and CGD are considered as differential diagnosis. The demonstration of successful patient-orientated treatment after using sequencing to confirm CGD and proper microbiological methods to identify the presence of specific infectious agents emphasises the importance of adopting this approach across the region.
Abbreviations
- BAL:
-
Bronchoalveolar lavage
- CGD:
-
Chronic granulomatous disease
- DHR:
-
Dihydrorhodamine-1,2,3
- GAM:
-
Galactomannan
- NAPDH:
-
Nicotinamide adenine dinucleotide phosphate
- NBT:
-
Nitroblue tetrazolium
- NCF1:
-
Neutrophil cytosolic factor 1
- PMA:
-
Phorbol 12-myristate 13-acetate
- PMN:
-
Polymorphonuclear neutrophils
- ROS:
-
Reactive oxygen species
References
Barlogis V, Suarez F, Lanternier F, et al. Epidemiology and outcome of invasive fungal diseases in patients with chronic granulomatous disease: a multicenter study in France. Pediatr Infect Dis J. 2011;30:57–62.
Blumental S, Mouy R, Mahlaoui N, Bougnoux ME, Debré M, Beauté J, Lortholary O, Blanche S, Fischer A. Invasive mold infections in chronic granulomatous disease: a 25-year retrospective survey. Clin Infect Dis. 2011;53:e159–69.
Marciano BE, Spalding C, Fitzgerald A, Mann D, Brown T, Osgood S, Yockey L, Darnell DN, Barnhart L, Daub J, et al. Common severe infections in chronic granulomatous disease. Clin Infect Dis. 2015;60:1176–83.
Meischl C, Roos D. The molecular basis of chronic granulomatous disease. Springer Semin Immunopathol. 1998;19:417–34.
Winkelstein JA, Marino MC, Johnston RB, Boyle J, Curnutte J, Gallin JI, Malech HL, Holland SM, Ochs H, Quie P. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore). 2000;79:155–69.
Roos D, Kuhns DB, Maddalena A, Roesler J, Lopez JA, Ariga T, Avcin T, de Boer M, Bustamante J, Condin-Neto M, et al. Hematologically important mutations: X-linked chronic granulomatous disease (third update). Blood Cells Mol Dis. 2010;45:246–65.
Roesler J, Hecht M, Freihorst J, Lohmann-Matthes ML, Emmendorffer A. Diagnosis of chronic granulomatous disease and of its mode of inheritance by dihydrorhodamine 123 and flow microcytofluorometry. Eur J Pediatr. 1991;150(3):161–5.
Buvelot H, Posfay-Barbe KM, Linder P, Schrenzel J, Krause KHStaphylococcus aureus, phagocyte NADPH oxidase and chronic granulomatous disease. FEMS Microbiol Rev. 2017;41(2):139-57.
Gabrielli E, et al. Osteomyelitis caused by aspergillus species: a review of 310 reported cases. Clin Microbiol Infect. 2014;20:559–65.
Vowells SJ, Sekhsaria S, Malech HL, Shalit M, Fleisher TA. Flow cytometric analysis of the granulocyte respiratory burst: a comparison study of fluorescent probes. J Immunol Methods. 1995;178:89–97.
Holland SM. Neutropenia and neutrophil defects. Manual of clinical laboratory immunology. Washington: ASM; 2002. p. 861–2.
Heshmatnia J, Marjani M, Mahdaviani SA, Adimi P, Pourabdollah M, et al. Paecilomyces formosus infection in an adult patient with undiagnosed chronic granulomatous disease. J Clin Immunol. 2017;37:342–6.
Vowells SJ, Fleisher TA, Sekhsaria S, Alling DW, Maguire TE, et al. Genotype-dependent variability in flow cytometric evaluation of reduced nicotinamide adenine dinucleotide phosphate oxidase function in patients with chronic granulomatous disease. J Pediatr. 1996;128:104–7.
Epling CL, Stites DP, McHugh TM, Chong HO, Blackwood LL, Wara DW. Neutrophil function screening in patients with chronic granulomatous disease by a flow cytometric method. Cytometry. 1992;13:615–20.
Nasri T, Hedayati MT, Abastabar M, Pasqualotto AC, Armaki MT, et al. Hoseinnejad a, Nabili M. , PCR-RFLP on β-tubulin gene for rapid identification of the most clinically important species of aspergillus. J Microbiol Methods. 2015;117:144–7.
Mirhendi H, Kambiz Diba K, Kordbacheh P, Nilufar Jalalizand N, Kuichi Makimura K. Identification of pathogenic aspergillus species by a PCR-restriction enzyme method. J Med Microbiol. 2007;56(11):1568–70.
Dekker J, de Boer M, Roos D. Gene-scan method for the recognition of carriers and patients with p47(phox)-deficient autosomal recessive chronic granulomatous disease. Exp Hematol. 2001;29:1319–25.
Iwen PC, Rupp ME, Langnas AN, Reed EC, Hinrichs SH, et al. Invasive pulmonary aspergillosis due to aspergillus terreus: 12-year experience and review of the literature. Clin Infect Dis. 1998;26:1092–7.
Schett G, Casati B, Willinger B, Weinländer G, Binder T, Grabenwöger F, Sperr W, Geissler K, Jäger U. Endocarditis and aortal embolization caused by Aspergillus terreus in a patient with acute lymphoblastic leukemia in remission: diagnosis by peripheral-blood culture. J Clin Microbiol. 1998;36(11):3347–51.
Tritz DM, Woods GL. Fatal disseminated infection with aspergillus terreus in immunocompromised hosts. Clin Infect Dis. 1993;16:118–22.
Nakahara Y, Katoh O, Yamada H, Sumida I, Hanada M. Allergic bronchopulmonary aspergillosis caused by aspergillus terreus presenting with lobar collapse. Intern Med. 1992;31:140–2.
Kimura M, Udagawa S, Shoji A, et al. Pulmonary aspergillosis due to aspergillus terreus combined with staphylococcal pneumonia and hepatic candidiasis. Mycopathologia. 1990;111:47–53.
Moore CK, Hellreich MA, Coblentz CL, Roggli VL. Aspergillus terreus as a cause of invasive pulmonary aspergillosis. Chest. 1988;94:889–91.
Chang SW, King TE. Aspergillus Terreus causing invasive pulmonary aspergillosis with air-crescent sign. J Natl Med Assoc. 1986;78(3):248 251-253.
Lass-Florl, Cornelia; Griff K.; Mayr A.; Petzer A.; Gastl G.; Bonatti H.; Freund M.; Kropshofer G.; Dierich M.; Nachbaur D ."Epidemiology and outcome of infections due to aspergillus terreus: 10-year single Centre experience". Br J Haematol2005; 131:20–207.
Glotzbach RE. Aspergillus terreus infection of pseudoaneurysm of aortofemoral vascular graft with contiguous vertebral osteomyelitis. Am J Clin Pathol. 1982;77:224–7.
Brown DL, Musher DM, Taffet GE. Hematogenously acquired aspergillus vertebral osteomyelitis in seemingly immunocompetent drug addicts. West J Med. 1987;147(1):84–5.
Grandière-Perez L, Asfar P, Foussard C, Chennebault JM, Penn P, Degasne I. Spondylodiscitis due to aspergillus terreus during an efficient treatment against invasive pulmonary aspergillosis. Intensive Care Med. 2000;26:1010–1.
Park KU, Lee HS, Kim CJ, Kim EC. Fungal discitis due to aspergillus terreus in a patient with acute lymphoblastic leukemia. J Korean Med Sci. 2000;15:704–7.
Maman E, Morin AS, Soussan M, Coignard H, Lortholary O, Fain O. Multifocal bone aspergillosis by aspergillus terreus in an apparently immunocompetent patient. Presse Med. 2015;44(10):1064–6.
Elsawy A, Faidah H, Ahmed A, Mostafa A, Mohamed F. Aspergillus terreus meningitis in immunocompetent patient: a case report. Front Microbiol. 2015;6:1353.
Baddley JW, Pappas PG, Smith AC, Moser SA. Epidemiology of aspergillus terreus at a university hospital. J Clin Microbiol. 2003;41:5525–9.
Blum G, Perkhofer S, Haas H, Schrettl M, Würzner R, Dierich MP, et al. Potential basis for amphotericin B resistance in aspergillus terreus. Antimicrob Agents Chemother. 2008;52:1553–5.
Azoulay É. Pulmonary involvement in patients with hematological malignancies. Pulmonary involvement in patients with hematological malignancies: Springer; 2011. p. 5–7.
Hachem R, Ribeiro Gomes MZ, El Helou G, El Zakhem A, Kassis C, Ramos E, Jiang Y, Chaftari A-M, Raad II. Invasive aspergillosis caused by Aspergillus terreus: an emerging opportunistic infection with poor outcome independent of azole therapy. J Antimicrob Chemother. 2014;69:3148–55.
Kathuria S, Sharma C, Singh PK, Agarwal P, Agarwal K, Hagen F, et al. Molecular epidemiology and in-vitro antifungal susceptibility of aspergillus terreus species complex isolates in Delhi, India: evidence of genetic diversity by amplified fragment length polymorphism and microsatellite typing. PLoS One. 2015;10(3):e0118997.
Pastor FJ, Guarro J. Treatment of aspergillus terreus infections: a clinical problem not yet resolved. Int J Antimicrob Agents. 2014;44(4):281–9.
Dagenais TR, Keller NP. Pathogenesis of aspergillus fumigatus in invasive aspergillosis. Clin Microbiol Rev. 2009;22(3):447–65.
Goh JG, Ravikumar S, Win MS, Cao Q, Tan AL, Lim JH, et al. Neutrophils differentially attenuate immune response to aspergillus infection through complement receptor 3 and induction of myeloperoxidase. Cell Microbiol. 2018;20(3):e12798.
Van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, et al. Chronic granulomatous disease: the European experience. PLoS One. 2009;4:5234.
Fattahi F, Badalzadeh M, Sedighipour L, Movahedi M, Fazlollahi MR, Mansouri SD, et al. Inheritance pattern and clinical aspects of 93 Iranian patients with chronic granulomatous disease. J Clin Immunol. 2011;31(5):792–801.
Funding
IMA is supported by the Wellcome Trust (093080/Z/10/Z) and by the Dunhill Medical Trust (R368/0714).
Availability of data and materials
All data obtained is presented in the manuscript. No additional datasets were generated or analyzed in the current study.
Author information
Authors and Affiliations
Contributions
EM, IMA and PT were involved in the study design and data collection and had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. EM, PT and SS wrote the initial draft of the manuscript. KvL and DR conducted the sequencing analysis. SS, MM, AM and EM conducted the clinical and laboratory tests. EM, SS, MM, AM, DM, PM, KvL, DR, JG, IMA and PT were involved in data analysis and interpretation, reviewing the manuscript and providing critical feedback and assessment. All authors have seen and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Patient consent for case report publication in journal and has signed consent form.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mortaz, E., Sarhifynia, S., Marjani, M. et al. An adult autosomal recessive chronic granulomatous disease patient with pulmonary Aspergillus terreus infection. BMC Infect Dis 18, 552 (2018). https://doi.org/10.1186/s12879-018-3451-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-018-3451-8