
Abstract
Background
Plant photosynthesis can be improved by elevated CO2 concentration (eCO2). In vitro growth under CO2 enriched environment can lead to greater biomass accumulation than the conventional in micropropagation. However, little is know about how eCO2 promotes transformation of grape plantlets in vitro from heterotrophic to autotrophic. In addition, how photosynthesis-related genes and their proteins are expressed under eCO2 and the mechanisms of how eCO2 regulates RbcS, Rca and their proteins have not been reported.
Results
Grape (Vitis vinifera L. cv. ‘Pinot Noir’) plantlets in vitro were cultured with 2% sucrose designated as control (CK), with eCO2 (1000 μmol·mol− 1) as C0, with both 2% sucrose and eCO2 as Cs. Here, transcriptomic and proteomic profiles associated with photosynthesis and growth in leaves of V. vinifera at different CO2 concentration were analyzed. A total of 1814 genes (465 up-regulated and 1349 down-regulated) and 172 proteins (80 up-regulated and 97 down-regulated) were significantly differentially expressed in eCO2 compared to CK. Photosynthesis-antenna, photosynthesis and metabolism pathways were enriched based on GO and KEGG. Simultaneously, 9, 6 and 48 proteins were involved in the three pathways, respectively. The leaf area, plantlet height, qP, ΦPSII and ETR increased under eCO2, whereas Fv/Fm and NPQ decreased. Changes of these physiological indexes are related to the function of DEPs. After combined analysis of proteomic and transcriptomic, the results make clear that eCO2 have different effects on gene transcription and translation. RbcS was not correlated with its mRNA level, suggesting that the change in the amount of RbcS is regulated at their transcript levels by eCO2. However, Rca was negatively correlated with its mRNA level, it is suggested that the change in the amount of its corresponding protein is regulated at their translation levels by eCO2.
Conclusions
Transcriptomic, proteomic and physiological analysis were used to evaluate eCO2 effects on photosynthesis. The eCO2 triggered the RbcS and Rca up-regulated, thus promoting photosynthesis and then advancing transformation of grape plantlets from heterotrophic to autotrophic. This research will helpful to understand the influence of eCO2 on plant growth and promote reveal the mechanism of plant transformation from heterotrophic to autotrophic.
Similar content being viewed by others

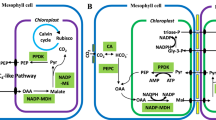

Background
Increasing atmospheric CO2 concentration influences plant growth [1, 2]. Photosynthesis, respiration and water relations are the three primary physiological processes influenced by elevated CO2 concentration (eCO2) in plants [3]. CO2 concentration inside the culture vessels decreased when plantlets grown in vitro, which limits the photosynthetic rate of the plants [4, 5]. The biomass accumulation of the in vitro cultured plants increased under photoautotrophic and CO2 enrichment conditions, also affected nutrient absorption and secondary metabolism [6, 7].
Plantlet grown vigorously under CO2 enriched photoautotrophic and photomixotrophic conditions, with high photosynthetic photon flux density [8]. Photosynthetic response to light and CO2 increased with Rubisco activities and proteins of plantlets grown in vitro [7]. Rubisco, the main catalytic enzyme determines photosynthetic rate [8], would respond to eCO2 [9] and increase carboxylation efficiency under eCO2 [10]. Succinctly, the synthesis of the Rubisco holoenzyme is mainly affected by ribulose bisphosphate carboxylase small chain (RbcS) [11]. The activity of Rubisco is related to Rubisco activase (Rca) and other proteins [12, 13].
In some species, it is reported that the transcript levels of RbcS are differentially regulated by red and blue light or growth temperature [14]. The abundance of the RbcS multigene family transcript has been researched in many plants [15]. RbcS regulates Rubisco through coordinated expression of RbcL and RbcS in plants [11]. In addition to the folded RbcL subunits assemble [16], RbcS could combine more CO2 than the RbcL in all Rubiscos [17]. The detailed mechanism of RbcS mediated assembly of RbcL under different environment and how the expression of RbcS and its protein responds to eCO2 remains to be investigated. The Rca could gain energy from ATP hydrolysis to remodel Rubisco inhibitors and activate Rubisco [18]. Inhibit expression of Rca in some plants results in severe photoautotrophic growth defects [19]. Rca proteins belong to a subgroup of the ATPases associated with various cellular activities (AAA) called AAA+ [20]. There are two Rca forms both can activate Rubisco [21]. Rca is regulated by the intracellular ATP/ADP ratio [22] or the C-terminal extension of the α-isoform of Rca in some plants [18]. Some research indicated that Rca could reduce the effects of abiotic stresses on plants, such as high temperature, drought, salt [23,24,25] and heavy metal [26]. The expression of Rca is regulated by trans-acting factors in soybean [27]. The actual change mechanism of Rca expression and whether Rca related to other proteins under eCO2 is less studied.
‘Pinot Noir’ is a wine grape variety widely planted in worldwide and its growth influenced by various environmental factors [28]. The increasing CO2 concentration could promote plant growth. Although, many studies have focused on the effects of CO2 on grape ripening [29] and postharvest [30]. It is unclear the mechanism of how eCO2 affects the plant growth and photosynthesis. Additionally, there are a few reports on the analysis of transcriptome combined with proteome to study the effects of eCO2 on grape growth and development. In light of this situation, the experiment was conducted based on the hypothesis that eCO2 will enhance photosynthesis by regulating the expression of related genes and proteins in grape plantlets. Therefore, grape plantlets grown in vitro cultured with eCO2 were used in this study based on transcriptome, proteome and photosynthetic physiology analysis.
Results
Effects of eCO2 on growth and chlorophyll fluorescence
Grape plantlets were cultured for 25 days at 1000 μmol·mol− 1 of CO2 and compare with control conditions. The results showed that the leaf area, plantlet height and shoot fresh weight increased significantly in Cs and C0 compared with CK (Additional file 1: Table S1). In addition, the number of adventitious roots in tubers also was increased in Cs and C0 (Fig. 1a).

Phenotypical characteristics and fluorescence parameter changes of ‘Pinot Noir’ plantlet in vitro caused by eCO2 (1000 μmol·mol− 1) on the 25th day. a Changes of the morphology and growth. b, c Response of optimal photochemical efficiency of PSII (Fv/Fm). d Changes of non-photochemical quenching (NPQ), photochemistry quenching (qP) and effective quantum yields of PSII (ΦPSII). eThe change of photosynthetic electron transport (ETR)
In compared to CK, the Fv/Fm decreased in Cs and C0, and significantly lower in C0 than that of CK (Fig. 1b, c). The qP and ETR rised in Cs and C0 (Fig. 1d, e). The ETR of C0 was significantly higher than the Cs and CK. The qP of C0 was significantly higher than Cs and CK (Fig. 1d). The NPQ was in the following order: Cs < C0 < CK (Fig. 1d). The decrease of NPQ indicated that eCO2 enhanced the efficiency of PSII and reduced the damage caused by biotic and abiotic stress. The ΦPSII of Cs and C0 were significantly higher than CK (Fig. 1d). These results suggested that eCO2 improved photosynthesis and reflected by chlorophyll fluorescence parameters, including Fv/Fm, ETR, qP, NPQ and ΦPSII.
Transcriptome and proteome differences expression in eCO2
In the transcriptome project, three RNA-Seq groups with three replications were sequenced, 29.5Gb clean bases were generated from the 9 libraries. After data processing, 46.49–47.46 million high-quality reads were obtained (Table 1). Through transcriptome analysis, a total of 1814 DEGs were observed by comparing with CK, of which 116 up-regulated and 632 down-regulated DEGs were identified in Cs versus CK, 349 up-regulated and 717 down-regulated DEGs were identified in C0 versus CK (Fig. 2a). According to SDS-PAGE analysis, protein sample could be tested in the next step (Additional file 2: Figure S1). After analysis of proteomic profiling, a total of 177 DEPs were observed from the pooled data for above two comparison groups. Among them, 48 up-regulated DEPs and 67 down-regulated DEPs were identified in Cs versus CK, 32 up-regulated DEPs and 30 down-regulated DEPs were identified in C0 versus CK (Fig. 2b).

Venn diagram showing of number of DEGs and DEPs expressed in different treatments. a The DEGs identified with Cs versus CK and C0 versus CK. b The DEPs identified with Cs versus CK and C0 versus CK
GO analysis of DEGs and DEPs
Of the 25,679 genes identified in the transcriptome analysis, 17,750 genes (69.12%) were annotated via GO analysis. Compared with CK, 748 DEGs identified in Cs were enriched in the biological process (BP), cellular component (CC), and molecular function (MF) categories. In the cellular components category, most of DEGs were involved in integral component of membrane (99 genes) and cytoplasm (94 genes). In the biological process category, most of DEGs were involved in defense response (70 genes) and transcription, DNA-templated (59 genes). In the molecular function category, most of DEGs were involved in transcription factor activity, sequence-specific DNA binding (54 genes) and ATP binding (48 genes) (Additional file 3: Table S2 A).
The 1066 DEGs of C0 versus CK were detected. Most DEGs mainly enriched in cytoplasm (98 genes) and integral component (97 genes) of cellular components. In the biological process category, most of DEGs were involved in defense response (53 genes) and transcription, DNA-templated (48 genes). In the molecular function category, most of DEGs were involved in metal ion binding (52 genes) and transcription factor activity, sequence-specific DNA binding (45 genes) (Additional file 3: Table S2 B).
From the pooled data for Cs versus CK, 115 DEPs were enriched in cell part (68 proteins) of cellular components. In the biological process category, most of DEPs were involved in metabolic process (81 proteins). In the molecular function category, most of DEPs were involved in catalytic activity (59 proteins) (Additional file 4: Table S3 A). Among the top 10 up-regulated DEPs, there were 3 proteins with abundance change related to photosynthesis: oxygen-evolving enhancer protein 3 (PsbQ; XP_002275624.1), chlorophyll a-b binding protein CP26 (Lhcb5; XP_002264295.1), photosystem II protein V (PsbE; YP_567093.1) (Table 2). But ribulose bisphosphate carboxylase/oxygenase activas (Rca; XP_002282979.1) was the 11th top up-regulated DEPs. Among the top 10 down-regulated DEPs, 2 proteins related to secondary metabolites: aspartokinase 2 (XP_010660689.1) and protein luteindeficient 5 (XP_002279984.3); one protein related to polysaccharide catabolic process: inactive beta-amylase 9 (XP_002276777.1); one protein related to stress tolerance: acid phosphatase 1 (XP_003632911.1) (Table 2).
A total of 62 DEPs were detected in C0 versus CK. DEPs were annotated and enriched in the three categories. In the cellular components, most proteins were involved in cell part (36 proteins). In the biological process category, most proteins were involved in metabolic process (45 proteins). In the molecular function category, most proteins were involved in catalytic activity (28 proteins) (Additional file 4: Table S3 B). Among the top 10 up-regulated DEPs, 5 of the top 10 DEPs were photosynthesis proteins: oxygen-evolving enhancer protein 3 (PsbQ; XP_002275624.1), Plastocyanin (Pc; XP_002285904.1), Chlorophyll a-b binding protein (Lhcb6; XP_002263201.1), chlorophyll a-b binding protein CP26 (Lhcb5; XP_002264295.1) and chlorophyll a-b binding protein of LHCII type 1 (Lhcb1; XP_002283566.1) (Table 3). The Rca (XP_002282979.1) was the 13th top up-regulated DEPs. Three of the top 10 DEPs down-regulated proteins: bifunctional 3-dehydroquinate dehydratase/shikimate dehydrogenase (XP_002270232.1, XP_002270188.1) and beta-glucosidase 13 (XP_002270422.2) (Table 3) were associated with biosynthesis of secondary metabolites.
KEGG pathway analysis for DEPs
To further investigate the plant reaction to eCO2, DEPs were identified by searching the KEGG database. The 115 DEPs of Cs were assigned to 52 KEGG pathways, and the top 5 pathways with the highest rich factor were photosynthesis-antenna proteins, metabolic pathways, biosynthesis of secondary metabolites, carbon metabolism, biosynthesis of amino acids (Additional file 5: Table S4 A). The 62 DEPs of C0 were assigned to 20 KEGG pathways, and the top 5 pathways with the highest rich factor were photosynthesis-antenna proteins, photosynthesis, metabolic pathways, phenylpropanoid biosynthesis, phenylalanine, tyrosine and tryptophan biosynthesis (Additional file 5: Table S4 B).
The common pathways with the highest rich factor of Cs versus CK and C0 versus CK were photosynthesis-antenna proteins, photosynthesis and metabolic pathways. Simultaneously, 9, 6 and 48 proteins were involved in the three pathways, respectively. Moreover, 12 proteins involved in metabolic pathway were overlaps with photosynthesis (Table 4).
There were 8 chlorophyll a-b binding proteins: Chlorophyll a-b binding protein (Lhcb6, XP_002263201.1), chlorophyll a-b binding protein 151 (Lhcb2; XP_002271687.1), photosystem I chlorophyll a-b-binding protein 3–1 (Lhcb3; XP_002273201.1), chlorophyll a-b binding protein 13 (Lhcb3; XP_002274150.2), chlorophyll a-b binding protein of LHCII type 1 (Lhcb1; XP_002275075.1), chlorophyll a-b binding protein 13 (Lhcb3; XP_002284493.1), chlorophyll a-b binding protein of LHCII type 1 (Lhcb1; XP_002285646.1) and chlorophyll a-b binding protein of LHCII type 1 (Lhcb1; XP_002283566.1) significantly up-regulated in Cs and C0 compared with those in CK, only 1 protein, Chlorophyll a-b binding protein (Lhcb3; XP_003633024.1) was descend in Cs (Fig. 3). There were 4 subunits of PSII: PsbQ (XP_002275624.1), PsbE (YP_567093.1), photosystem II protein D2 (PsbD; YP_567071.1) and photosystem I reaction center subunit N (PsaN; XP_003631913.1) significantly up-regulated in Cs and C0, but 1 subunit Pc (XP_002285904.1) of PSI was down-regulated (Fig. 3). Ribulose bisphosphate carboxylase small chain (RbcS; XP_002276967.1) and ATP synthase delta chain (XP_002274963.1) were up-regulated in Cs and C0 (Figs. 3, 4). Other proteins: beta-glucosidase 13 (XP_002270422.2), beta-amylase 1 (XP_002276777.1), threonine dehydratase (XP_002264311.2) and GDP-L-galactose phosphorylase 2 (XP_002278339.1) involved in metabolic were down-regulated in Cs and C0 compared with those in CK, and also involved in polysaccharide catabolic process and biosynthesis of secondary metabolites (Table 4).

The DEPs and their corresponding genes expression of photosynthesis system

The expression of DEPs and their corresponding genes in formation of Rubisco
Combined analysis of transcriptome and proteome data
To reveal eCO2 regulates photosynthesis gene via transcript and protein levels, the transcript data were used to analyze 18 DEPs associated with photosynthesis and metabolic pathways. Seven DEPs: Lhcb6 (XP_002263201.1), Lhcb3 (XP_002273201.1, XP_002284493.1), Lhcb1 (XP_002275075.1, XP_002285646.1, XP_002283566.1) and RbcS (XP_002276967.1) and their mRNA expression showed up-regulated in Cs and C0. However, there were 3 DEPs, PsbQ (XP_002275624.1), Rca (XP_002282979.1) and Lhcb5 (XP_002264295.1) involved in photosynthesis up-regulated in Cs and C0, but their mRNA down-regulated. Other 8 DEPs were not correlated with genes expression under eCO2 (Figs. 3, 4).
The RbcS (XP_002276967.1) and its corresponding gene were up-regulated in Cs and C0. ATP synthase delta chain (XP_002274963.1) and Rca (XP_002282979.1) were up-regulated but their corresponding genes were down-regulated in Cs and C0 (Figs. 3, 4). The results make clear that eCO2 have different effects on gene transcription and translation. RbcS was not correlated with its mRNA level, suggesting that the change in the amount of RbcS is regulated at their transcript levels by eCO2. However, Rca was negatively correlated with its mRNA level, it is suggested that the change in the amount of its corresponding protein is regulated at their translation levels by eCO2.
Confirmation of qRT-PCR
In order to evaluate our transcriptome-sequencing data, 18 genes in the photosynthesis and metabolic pathway were selected for qRT-PCR. The results analyse indicated that 15 genes (83.33%) showed similar trends in the relative expression levels, which suggested that the gene expression changes detected by transcriptome-sequencing analysis were reliable. But 3 genes (16.67%) analyzed by qRT-PCR, i.e., PsbE (4025054), PsbD (4025083) and LHCB3 (LOC100252004) were not consistent with our RNA-seq data (Fig. 5).

qRT-PCR validation of the relative expression levels of 18 slected genes from Cs, C0 and CK in leaves
Discussion
Proteins involved in photosynthesis were regulated by eCO2
Light-harvesting complexes (LHC) of photosynthetic plant bind pigments essential for augmenting light capture and photoprotection [31]. LHCI and LHCII belong to photosystem I (PSI) and photosystem II (PSII), respectively. LHCII is a trimeric light-harvesting complex (Lhc) composed of a combination of the Lhcb gene products and others [32]. Plants can develop strategies of acclimating varies light conditions during seasons and can rapidly adjust photosynthesis antenna sizes in case of excess light, avoiding over excitation and formation of harmful by products [33]. CO2 concentration could affect the primary light reaction of photosynthesis in soybean leaves [34]. In our research, 8 proteins of LHCII were up-regulated in eCO2 (Table 4), this change indicated that eCO2 could induce more light-harvesting proteins (Fig. 3), and cause an increase in the size of the PSI and PSII antenna. The light-harvesting complex II (LHCII) could convert most photons to biochemical energy and biomass [35]. With the increase of LHCII in eCO2, more light energy can be absorbed and converted into photosystem. The increase of qP and decrease of NPQ confirming that leaves could absorb more light energy under eCO2 (Fig. 1d). In present study, the number of up-regulated light-harvesting proteins of PSII was more than that of PSI, which showed that the eCO2 had a great influence on PSII. Additionally, the LHCII conditions would migration from PSII to PSI under deficient CO2 environment [36]. CP24 was up-regulated in eCO2, it was essential for connecting LHCII to the PSII complex [37, 38]. Lack of the light-harvesting complex CP24 affects the structure and function of the grana membranes of higher plant chloroplasts [37]. Overall, these proteins, which were up-regulated under eCO2, could absorb and convert more light energy into the photosystem.
In photosynthesis pathway, the expression of PetE (XP_002285904.1) and Chlorophyll a-b binding protein (XP_003633024.1) were descend in Cs. Interestingly, the expression of PetE (LOC100248911) and LHCB3 (LOC100252004) were ascend in Cs and C0. These results indicated that most of the DEPs and their corresponding genes expression were inconsistent. The eCO2 may cause various modifications of related proteins after translation and needs to be study for further.
eCO2 regulates metabolic protein expression
There were 48 DEPs involved in metabolic pathway, while 12 of them were overlaps with photosynthesis. This might indicate that eCO2 would affect other metabolic through adjusting photosynthesis. Our results indicated many of down-regulated DEPs were enriched in metabolic pathway in eCO2, which were related to biosynthesis of secondary metabolites (Table 4). This change is suggesting that eCO2 probably decreased biosynthesis of secondary metabolites [39]. Therefore, plant could accumulation more primary metabolism products to encourage growth.
The eCO2 could ameliorate the effects caused by drought [40], high temperatures [41], and maintaining higher photosynthetic rates. This may be linked to the reduction in stomatal conductance [42]. Moreover, increasing photosystem antenna size must inevitably cause structural changes needed to ensure high efficiency of its functioning [43]. There were 4 DEPs (PsbQ, PsbE, PsbD and PsaN; Fig. 3) up-regulated in eCO2. Those proteins could maintain the stability of the photosystem reaction center [44, 45]. By analyzing changes of those proteins in eCO2, we can conclude that eCO2 could trigger some proteins to maintain the stability of the photosynthesis system. Therefore, eCO2 could ameliorate the adverse effect under abiotic stress. PsbQ can increase PSII activity and stability of oxygen release complexes (OECs) [45]. It is also the water decomposition subunit [46]. The PsbQ was up-regulated in eCO2, this means eCO2 could promote water decomposition and maintain stability in OECs by regulating PsbQ. The other 3 proteins (PsbE, PsbD and PsaN) related to photosynthetic electron transport and accumulation of photosynthetic substances [44]. Those proteins (LHCs, PsbQ, PetE, PsbD, PsaN) increased in eCO2 (Fig. 3), resulting in absorbing more light energy and promoting more photosynthetic electron transport. This is causing the advance of qP and ETR, and the reduction of NPQ (Fig. 1d, e).
ATP synthase delta chain is CF1 subunit (δ) belongs to the F-type ATPase, which utilizes the energy of a transmembrane electrochemical gradient to generate ATP by rotary catalysis [47]. F-type ATPase products would provide energy for photosynthesis carbon fixation [48]. ATP synthesis in the hydrophilic α3β3 head (CF1) is powered by the CF0 rotary motor in the membrane [49]. Previous studies have shown that the ATP synthase delta chain is mainly related to the component linkage of the F-type ATPase sector [50, 51]. In our study, ATP synthase delta chain protein was up-regulated in eCO2 (Fig. 3), this indicated that eCO2 can affect leaf redox pathways by changing the F-type ATPase subunit accumulation. Our results confirmed that ATP synthase delta chain act as a stator to prevent unproductive rotation of CF1 with CF0, this is consistent with previous study [49].
eCO2 promotes up-regulation of RbcS and Rca
Rubisco is L8S8 hexadecamer complex [52] and inefficient [53]. RbcS regulates Rubisco through coordinated expression of RbcL and RbcS in plants [11]. RbcS is linked to the folded RbcL subunits assemble [54] and as a ‘reservoir’ for CO2 storage [17]. In our results, RbcS was up-regulated in eCO2 (Fig. 4), this indicated that RbcS not only has high affinity with CO2, but also responds to eCO2 in the environment. It has reported that RbcS mRNA levels and RbcS synthesis simultaneously increased in RbcS-sense plants [11]. The RbcS transcript was found to be inhibited in source of sugar (sucrose or glucose) in the media of photoautotrophic Chenopodium callus and some plants, but over-expression of RbcS was found in low CO2 [55]. Interestingly, RbcS mRNA level was up-regulated in C0, and down-regulated in Cs and CK, which indicate that the medium with sugar inhibits the expression of RbcS, this is consistent with previous studies. The amount of RbcS synthesize was tightly correlated with RbcL mRNA level [11]. In our research, large amounts of RbcS accumulated under eCO2 but there was no significant change in RbcL mRNA level. This result showed that RbcS accumulated not only associated with RbcL mRNA level, but also related to CO2 concentration. It has reported that long-term growth of Arabidopsis at high CO2 (1000 μmol·mol− 1) resulted in nonstructural carbohydrates increased and an even greater decline in mRNA of RbcS [56]. Nevertheless, the mechanism of eCO2 regulates RbcS accumulated would research in future.
Sugar phosphate inhibits Rubisco activity [57], such as RuBP, CATP and Xu5P [12]. Rca catalyzes the remodeling of inactive Rubisco, releases it’s bound sugar phosphate and activate Rubisco [20]. Heat [23], drought [24] and salt [25] could increase Rca. In our results, Rca was up-regulated under eCO2. Through the previous analysis, LHCII, PsbQ, PsbE, PsbD, PsaN and ATP synthase delta chain were up-regulated, indicating that these proteins would absorb more energy and produce more ATP, which could change ATP/ADP ratio. Rca uses the hydrolysis of ATP to facilitate the dissociation of RuBP bound as an inhibitor at the active site of uncarbamylated and inactive Rubisco [58]. Therefore, the activity of Rca was affected by ADP/ATP ratio [22]. We speculated that eCO2 affect Rca activity by up-regulating the expression of light-harvesting proteins and F-type ATPase, and all of those changes ultimately affect the activation of Rubisco. Galmés et al. [59] reported that Rubisco content reduced was the primary driver in the regulation of Rubisco activity to eCO2. At normal conditions, Rca negatively affects the Rubisco content [60]. However, Rca level is a major limiting factor of non-steady-state photosynthesis [61]. Therefore, Rca up-regulated to adjust the non-steady-state photosynthesis caused by eCO2. Overproduction of Rubisco does not enhance photorespiration as well as CO2 assimilation probably due to partial deactivation of Rubisco [62]. Rca was negatively correlated with mRNA levels, it is suggested that changes in the expression of these proteins are regulated at their translation levels by eCO2.
Conclusions
The detailed analysis of transcriptome and proteome of grape (V. vinifera L. cv. ‘Pinot Noir’) plantlets in vitro under differential concentration of CO2 revealed crucial molecular mechanism difference in transformation from heterotrophic to autotrophic. The results indicated that eCO2 triggers the RbcS and Rca up-regulated, then promoting photosynthesis and then advancing transformation of grape plantlets from heterotrophic to autotrophic. The study provided deep refinements into the existing knowledge of plantlets in vitro response to eCO2, and the molecular mechanism was revealed through identification and comparative analysis of genes and proteins from photosynthesis-antenna, photosynthesis and metabolism pathways. The expression level of RbcS was not related to protein expression and the expression of Rca was highly inverse correlated with protein expression. Consequently, these datas provide clues as to the fundamental regulatory network targeted by eCO2, and will lead to future functional analyses that may be valuable for both agronomic improvement and our understanding of the means by which new phenotypes may arise.
Methods
Plant materials
‘Pinot Noir’ (V. vinifera L.) grape plantlets, which was kept in the Fruit Tree Physiology and Biotechnology Laboratory, College of Horticulture, Gansu Agricultural University, were used as test materials in an in vitro experiment. The grape plantlets were propagated in advance and were vigorous in growth without contamination. Each nodal segment (approximately 2.0 cm long) with two bud was cultured on modified B5 solid medium + IAA (0.1 mg·L− 1) (50 mL of medium was taken in 150 mL Erlenmeyer flasks). Plantlets were grown in controlled climate chamber (PQX-430D) at a day/night regime of 16 h/8 h (light/dark), an irradiance of 120 μmol·m− 2·s− 1, temperatures of 26 °C day and night. One climate chamber (PQX-430D-CO2) have TC-5000 (T) intelligent CO2 controller to regulate CO2 concentration. The CO2 concentration treatments were as follows: environmental atmospheric CO2 concentrations (380 ± 40 μmol·mol− 1); and elevated CO2 concentrations (1000 μmol·mol− 1). The grape plantlets were cultured with 2% sucrose designated as control (CK), with eCO2 while without sucrose as C0, with both 2% sucrose and eCO2 as Cs. Each treatment had three biological replicates with 15 plantlets per replicate. Plantlet leaves were harvested at 25 days after inoculation. The leaf samples were transferred immediately to liquid nitrogen and stored at − 80 °C for subsequent analysis. Different treatments were simultaneously sampled from three comparable plants used as three biological replications.
Chlorophyll fluorescence parameters
Chlorophyll fluorescence parameters of functional leaves were measured using the IMAG-PAM fluorometer (MAXI Imaging-PAM, Walz, Effeltrich, Germany). All daytime measurements were carried out between 10:00 and 12:00 in the morning. After the dark adaptation, minimal fluorescence (F0), steady fluorescence (Fs) and maximum fluorescence (Fm) were respectively measured under light irradiation (0.1110 and 2700 μmol·m− 2·s− 1). The optimal photochemical efficiency of PSII (Fv/Fm), effective quantum yields of PSII (ΦPSII), photochemistry quenching (qP) and photosynthetic electron transport (ETR) were calculated according to previous equations [63].
RNA isolation and library preparation for transcriptome analysis
Total RNA samples were extracted using the mirVana miRNA Isolation Kit (Ambion). The RNA samples were evaluated using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), with RNA Integrity Number (RIN) ≥ 7 were subjected to the subsequent analysis. The libraries were constructed using TruSeq Stranded mRNA LTSample Prep Kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. Then these libraries were sequenced on the Illumina sequencing platform (HiSeqTM 2500 or Illumina HiSeq X Ten) and 125 bp/150 bp paired-end reads were generated.
Analysis of RNA-sequencing data
Raw data (raw reads) were filtered into clean reads using NGS QC Toolkit. The reads containing ploy-N and the low quality reads were removed to obtain the clean reads. Then the clean reads were mapped to reference genome sequence (http://www.genoscope.cns.fr/externe/GenomeBrowser/Vitis/) using HISTA 2. Briefly, the number of mapped reads for each transcript was normalized into a reads per kb per million reads value (RPKM) to calculate level of differential expression for each transcript. In analysis, a criterion of P value < 0.05 and fold change > 2 or fold change < 0.5 was used to identify DEG. Functional gene classification was performed using UniProtKB/Swiss-Prot database. GO enrichment and KEGG pathway enrichment analysis of DEGs were performed using the R programming language based on the hypergeometric distribution, respectively.
qRT-PCR analysis
One micrograms total RNA was subjected to reverse transcription using SYBR Green PCR Master Mix (TaKaRa) Kit with gDNA Eraser (Perfect for Real Time). Real-time PCR was carried out by using SYBRs Premix Ex Taq II (TaKaRa) in ABI StepOne™ Plus Real-Time PCR System (Roche, Switzerland). All primers used for qRT-PCR were listed in Additional file 6: Table S5.
Protein extraction
Fresh leaves (0.5 g) from each biological replicate were ground into power in liquid nitrogen and dissolved (vortex blending) with 500 μL extraction buffer (0.7 M sucrose, 0.1 M NaCl, 0.5 M Tris-HCl (pH 7.5), 50 mM EDTA and 0.2% DTT). The samples were grinded at the power of 60 Hz for 2 min. Then supplemented with extraction buffer for 1 mL and mixed and added with Tris-phenol buffer and mixed for 30 min at 4 °C. The mixtures were centrifuged at 7100 g for 10 min at 4 °C. Collect phenol supernatants and added for 5 volumes of 0.1 M cold ammonium acetate-methanol buffer and precipitated at − 20 °C overnight. The samples were centrifuged at 12,000 g for 10 min to collect precipitations. The precipitations were dried and dissolved in lysis buffer (1% DTT, 2% SDS, 10% glycerinum, 50 mM Tris-HCl (pH 6.8) for 3 h. The samples were centrifuged at 12000 g for 10 min to collect supernatants. The supernatants were centrifuged again to remove precipitations completely. The protein concentration was quantified by BCA method [64] and theprotein purity was detected by SDS-PAGE [65], 15μg proteins of each sample were separated on 12% SDS-PAGE gel.
Protein digestion and iTRAQ labeling
Protein digestion was performed according to the FASP procedure [66]. Brifely, protein sample (100 μg) was subjected with 120 μL reducing buffer (10 mM DTT, 8 M Urea, 100 mM TEAB, pH 8.0) on 10 K ultrafiltration tube and the solution was incubated at 60 °C for 1 h. IAA was added to the solution with the final concentration of 50 mM in the dark at room temperature for 40 min. The solutions were centrifuged on the filters at 12,000 g for 20 min at 4 °C. Remove the supernatant and add TEAB (100 μL, 100 mM) to the solutions and centrifuged at 12,000 g for 20 min. Collection the filter units into new tubes, add TEAB (100 μL, 100 mM) and followed with 2 μL sequencing-grade trypsin (1 μg·μL− 1), incubated for digestion at 37 °C for 12 h. The collections of digested peptides were centrifuge at 12,000 g for 20 min. The solutions were collected and lyophilized. The lyophilized samples were resuspended in TEAB (100 μL, 50 mM) and 40 μL of each sample was transferred into new tubes for labeling. Each sample add iTRAQ label reagent (iTRAQ® Reagents-8plex kit, Sigma) following the manufacturer’s protocol (Applied Biosystems, Foster City, CA, USA). All labeled peptides were pooled together.
RP chromatography separation
iTRAQ labeled peptides were fractionated by RP chromatography separation using the 1100 HPLC System (Agilent). RP separation was performed on the Agilent Zorbax Extend RP column (5 μm, 150 mm × 2.1 mm). Mobile phases A (2% acetonitrile in HPLC water) and B (98% acetonitrile in HPLC water) were used for RP gradient. The solvent gradient was set as follows: 0~8 min, 98% A; 8.00~8.01 min, 98%~ 95% A; 8.01~38 min, 95%~ 75% A; 38~50 min, 75~60% A; 50~50.01 min, 60~10% A; 50.01~60 min, 10% A; 60~60.01 min, 10~98% A; 60.01~65 min, 98% A. Tryptic peptides were separated at an eluent flow rate of 300 μL·min− 1 and monitored at 210 and 280 nm. Dried samples were harvested from 8 min to 50 min and elution buffer were collected in every minute and numbered from 1 to 10 with pipeline. The separated peptides were lyophilized for MS detection.
Mass spectrometry analysis
All LC-MS/MS analyses were performed on a Q-Exactive mass spectrometer (Thermo, USA) equipped with a Nanospray Flex source (Thermo, USA). The peptides mixtures were loaded by a capillary C18 trap column (3 cm × 100 μm, C18, 3 μm, 150 Å) and separated by a C18 column (15 cm × 75 μm, C18, 3 μm, 120 Å) on an ChromXP Eksigent system (AB Sciex). The flow rate was 300 nL·min− 1 and linear gradient was 70 min (0~0.5 min, 95%~ 92% A; 0.5~48 min, 92%~ 74% A; 48~61 min, 74%~ 62% A; 61~61.1 min, 62%~ 15% A; 61.1~67 min, 15% A; 67~67.1, 15%~ 95% A; 67.1~70 min, 95% A. mobile phase A = 2% ACN/0.1% FA and B = 95% ACN/0.1% FA). Full MS scans were acquired in the mass range of 300–1600 m/z with a mass resolution of 70,000 and the AGC target value was set at 1,000,000. The 10 most intense peaks in MS were fragmented with higher-energy collisional dissociation (HCD) with collision energy of 30. MS/MS spectra were obtained with a resolution of 17,500 with an AGC target of 200,000 and a max injection time of 50 ms. The Q-E dynamic exclusion was set for 15.0 s and run under positive mode.
Protein identification and function annotation
Raw data of iTRAQ-labeled proteins by was search against V. vinifera (Grape) genome protein database in National Center for Biotechnology Information (NCBI) using the Proteome DiscovererTM 2.2 (Thermo, USA). Database searches were performed with trypsin digestion specificity, and the cysteine alkylation was considered as parameters in the database searching. For protein quantification method, iTRAQ8-plex was selected. For protein identification, a decoy database search approach was used to determine the false discovery rate (FDR) with acceptance if their FDR < 1.0% while protein identification containing at least two peptides.
The molecular functions of the identified proteins were classified according to their gene ontology annotations and their biological functions. Only the proteins identified with at least two different peptides and P value < 0.05, and quantified with a ratio of fold change > 1.4 or fold change < 5/7 and P value < 0.05, were considered. The NCBI and Uniprot databases were chosen to the validation and annotation of the protein sequences. Gene Ontology (GO) annotation for the identified proteins was assigned according to Uniprot database (http://www.uniprot.org).
Statistical analysis
The control and treatment groups were analyzed for statistical significance of differences between multiple groups using one-way ANOVA followed by Duncan’s multiple comparisons test. All calculations were performed using SPSS software (version 21; IBM, Armonk, NY, USA). All results are presented as mean ± SD from 3 independent biological replications. Treatment means were separated by the Duncan multiple range test at P value less than 0.01. We use min-max normalization method through the R programming language (3.4.3, pheatmap) to analysis transcriptional and proteomic represent expression values of heat map.
Abbreviations
- DEGs:
-
Differentially expressed genes;
- DEPs:
-
Differentially expressed proteins
- eCO2 :
-
Elevated CO2 concentration
- GO:
-
Gene ontology
- iTRAQ:
-
Isobaric Tag for Relative Absolute Quantitation
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- qRT-PCR:
-
Quantitative real-time PCR
- RbcS:
-
Ribulose bisphosphate carboxylase small chain
- Rca:
-
Rubisco activase
- Rubisco:
-
Ribulose-1, 5-bisphosphate carboxylase/oxygenase
References
Bezemer TM, Jones TH. The effects of CO2 and nutrient enrichment on photosynthesis and growth of Poa annua in two consecutive generations. Ecol Res. 2012;27(5):873–82.
Usuda H. Effects of elevated CO2 on the capacity for photosynthesis of a single leaf and a whole plant, and on growth in a radish. Plant & Cell Physiology. 2006;47(2):262–9.
Dusenge ME, Duarte AG, Way DA. Plant carbon metabolism and climate change: elevated CO2 and temperature impacts on photosynthesis, photorespiration and respiration. New Phytol. 2018;221(1):32-49.
Park SY, Moon HK, Murthy HN, Kim YW. Improved growth and acclimatization of somatic embryo-derived Oplopanax elatus plantlets by ventilated photoautotrophic culture. Biol Plant. 2011;55:559–62.
Xiao Y, Niu G, Kozai T. Development and application of photoautotrophic micropropagation plant system. Plant Cell Tissue Organ Cult. 2011;105:149–58.
Arigita L, Canãl J, Tamés RS, González A. CO2-enriched microenvironment affects sucrose and macronutrients absorption and promotes autotrophy in the in vitro culture of kiwi (Actinidia deliciosa Chev. Liang and Ferguson). In Vitro Cell Dev Biol-Plant. 2010;46:312–22.
Tomimatsu H, Tang Y. Effects of high CO2 levels on dynamic photosynthesis: carbon gain, mechanisms, and environmental interactions. J Plant Res. 2016;129(3):365-377.
Parry MA, Andralojc PJ, Scales JC, Salvucci ME, Carmo-Silva AE, Alonso H, Whitney SM. Rubisco activity and regulation as targets for crop improvement. J Exp Bot. 2013;64(3):717-30.
Ainsworth EA, Rogers A. The response of photosynthesis and stomatal conductance to rising [CO2]: mechanisms and environmental interactions. Plant Cell Environ. 2007;30(3):258–70.
Gamage D, Thompson M, Sutherland M, Hirotsu N, Makino A, Seneweera S. New insights into the cellular mechanisms of plant growth at elevated atmospheric carbon dioxide concentrations. Plant Cell Environ. 2018;41(6):1233–46.
Suzuki Y, Makino A. Availability of rubisco small subunit up-regulates the transcript levels of large subunit for stoichiometric assembly of its holoenzyme in rice. Plant Physiol. 2012;160(1):533–40.
Chen J, Wang P, Mi HL, Chen GY, Xu DQ. Reversible association of ribulose-1, 5-bisphosphate carboxylase/oxygenase activase with the thylakoid membrane depends upon the ATP level and pH in rice without heat stress. J Exp Bot. 2010;61(11):2939–50.
Vitlin Gruber A, Feiz L. Rubisco assembly in the chloroplast. Front Mol Biosci. 2018;5:24.
Yoon M, Putterill JJ, Ross GS, Laing WA. Determination of the relative expression levels of rubisco small subunit genes in Arabidopsis by rapid amplification of cDNA ends. Anal Biochem. 2001;291(2):237–44.
Suzuki Y, Kihara-Doi T, Kawazu T, Miyake C, Makino A. Differences in rubisco content and its synthesis in leaves at different positions in Eucalyptus globulus seedlings. Plant Cell Environ. 2010;33(8):1314–23.
Joshi J, Mueller-Cajar O, Tsai YC, Hartl FU. Hayerhartl M (2015) role of small subunit in mediating assembly of red-type form I rubisco. J Biol Chem. 2015;290(2):1066–74.
Van LM, Hub SJ, Dsd V, Andersson I. CO2 and O2 distribution in rubisco suggests the small subunit functions as a CO2 reservoir. J Am Chem Soc. 2014;136(8):3165–71.
Portis AR Jr, Li CS, Wang DF, Salvucci ME. Regulation of rubisco activase and its interaction with rubisco. J Exp Bot. 2008;59:1597–604.
Mueller-Cajar O, Stotz M, Bracher A. Maintaining photosynthetic CO2 fixation via protein remodelling: the rubisco activases. Photosynth Res. 2014;119:191–201.
Bracher A, Whitney SM, Hart FU, Hayerhartl M. Biogenesis and metabolic maintenance of rubisco. Annu Rev Plant Biol. 2017;68(1):29.
Shen JB, Orozco EM Jr, Ogren WL. Expression of the two isoforms of spinach ribulose 1,5-bisphosphate carboxylase activase and essentiality of the conserved lysine in the consensus nucleotide-binding domain. J Biol Chem. 1991;266(14):8963–8.
Noji H, Ueno H, McMillan DGG. Catalytic robustness and torque generation of the F1-ATPase. Biophys Rev. 2017;9(2):103–18.
Wang D, Li XF, Zhou ZJ, Feng XP, Yang WJ, Jiang DA. Two rubisco activase isoforms may play different roles in photosynthetic heat acclimation in the rice plant. Physiol plantarum. 2010;139(1):55–67.
Ji K, Wang Y, Sun W, Lou Q, Mei H, Shen S, Chen H. Drought-responsive mechanisms in rice genotypes with contrasting drought tolerance during reproductive stage. J Plant Physiol. 2012;169(4):336–44.
Chen Y, Wang XM, Zhou L, He Y, Wang D, Qi YH, Jiang DA. Rubisco activase is also a multiple responder to abiotic stresses in rice. PLoS One. 2015;10(10):0140934.
Son JA, Narayanankutty DP, Roh KS. Influence of exogenous application of glutathione on rubisco and rubisco activase in heavy metal-stressed tobacco plant grown in vitro. Saudi J Biol Sci. 2014;21(1):89–97.
Zhang J, Du H, Chao M, Yin Z, Yang H, Li Y, Huang F, Yu D. Identification of two bZIP transcription factors interacting with the promoter of soybean rubisco activase gene (GmRCAα). Front Plant Sci. 2016;7:e0140934.
Kizildeniz T, Mekni I, Santesteban H, Pascual I, Morales F, Irigoyen JJ. Effects of climate change including elevated CO2 concentration, temperature and water deficit on growth, water status, and yield quality of grapevine (vitis vinifera L.) cultivars. Agr Water Manage. 2015;159:155–64.
Parra CS, Aguirreolea J, Sánchez-Díaz M, Irigoyen JJ, Morales F. Effects of climate change scenarios on Tempranillo grapevine (Vitis vinifera L.) ripening: response to a combination of elevated CO2 and temperature, and moderate drought. Plant Soil. 2010;337(1–2):179–91.
Rosales R, Romero I, Fernandez-Caballero C, Isabel Escribano M, Merodio C, Sanchez-Ballesta MT. Low temperature and short-term high-CO2 treatment in postharvest storage of table grapes at two maturity stages: effects on transcriptome profiling. Front Plant Sci. 2016;7(845):1020.
Boldt L, Yellowlees D, Leggat W. Hyperdiversity of genes encoding integral light-harvesting proteins in the dinoflagellate Symbiodinium sp. PLoS One. 2012;7(10):e47456.
Sandona D, Croce R, Pagano A, Crimi M, Bassi R. Higher plants light harvesting proteins. Structure and function as revealedbymutation analysis of eitherprotein or chromophore moieties. Biochim. Biophys. Acta. 1998;1365:207–14.
Pandit A, Van IS, Van HA, Croce R. Introduction: light harvesting for photosynthesis. Photosynth Res. 2018;135(1–3):1–2.
Ye ZP, Kang HJ, Duan SH, Wang YJ. Photosynthetic physio-ecological characteristics in soybean leaves at different CO2 concentrations. Chin J Appl Ecol. 2018;29(2):583–91.
Pietrzykowska M, Suorsa M, Semchonok DA, Tikkanen M, Boekema EJ, Aro EM, Janssona S. The light-harvesting chlorophyll a/b binding proteins Lhcb1 and Lhcb2 play complementary roles during state transitions in Arabidopsis. Plant Cell. 2014;26(9):3646.
Ueno Y, Shimakawa G, Miyake C, Akimoto S. Light-harvesting strategy during CO2-dependent photosynthesis in the green alga chlamydomonas Reinhardtii. J Phys Chem Lett. 2018;95:1028–33.
Kovacs L, Damkjaer J, Kereiche S, Ilioaia C, Ruban AV, Boekema EJ, Jansson S, Horton P. Lack of the light-harvesting complex CP24 affects the structure and function of the grana membranes of higher plant chloroplasts. Plant Cell. 2006;18(11):3106–20.
Bianchi SD, Dall'Osto L, Tognon G, Morosinotto T, Bassi R. Minor antenna proteins CP24 and CP26 affect the interactions between photosystem II subunits and the electron transport rate in grana membranes of Arabidopsis. Plant Cell. 2008;20(4):1012–28.
Johnson SN, Barton AT, Clark KE, Gregory PJ, Mcmenemy LS. Elevated atmospheric CO2 impairs the performance of root-feeding vine weevils by modifying root growth and secondary metabolites. Glob Chang Biol. 2011;17(2):688–95.
De Souza AP, Cocuron JC, Garcia AC, Alonso AP, Buckeridge MS. Changes in whole-plant metabolism during the frain-filling stage in sorghum grown under elevated CO2 and drought. Plant Physiol. 2015;169(3):1755–65.
Salazar-Parra C, Aranjuelo I, Pascual I, Erice G, Sanz-Sáez Á, Aguirreolea J, Sánchez-Díaz M, Irigoyen JJ, Araus JL, Morales F. Carbon balance, partitioning and photosynthetic acclimation in fruit-bearing grapevine (Vitis vinifera L. cv. Tempranillo) grown under simulated climate change (elevated CO2, elevated temperature and moderate drought) scenarios in temperature gradient greenhouses. J Plant Physiol. 2015;174:97–109.
Markelz RJ, Strellner RS, Leakey AD. Impairment of C4 photosynthesis by drought is exacerbated by limiting nitrogen and ameliorated by elevated [CO2] in maize. J Exp Bot. 2011;62(9):3235–46.
Taisova AS, Yakovlev AG, Fetisova ZG. Size variability of the unit building block of peripheral light-harvesting antennas as a strategy for effective functioning of antennas of variable size that is controlled in vivo by light intensity. Biochemistry Biokhimiia. 2014;79(3):251–9.
Kiss E, Kós PB, Chen M, Vass I. A unique regulation of the expression of the psbA, psbD, and psbE genes, encoding the D1, D2 and cytochrome b559 subunits of the photosystem II complex in the chlorophyll d containing cyanobacterium Acaryochloris marina. Biochim Biophys Acta. 2012;1817(7):1083–94.
Ido K, Nield J, Fukao Y. Cross-linking evidence for multiple interactions of the PsbP and PsbQ proteins in a higher plant photosystem II supercomplex. J Biol Chem. 2014;289(29):20150–7.
Haniewicz P, Floris D, Farci D, Kirkpatrick J, Loi MC, Büchel C, Bochtler M, Piano D. Isolation of plant photosystem II complexes by fractional solubilization. Fron Plant Sci. 2015;6:1100.
Allegretti M, Klusch N, Mills DJ, Vonck J, Kühlbrandt W, Davies KM. Horizontal membrane-intrinsic α-helices in the stator a-subunit of an F-type ATP synthase. Nature. 2015;521(7551):237–40.
Hisabori T, Sunamura EI, Kim Y, Konno H. The chloroplast ATP synthase features the characteristic redox regulation machinery. Antioxid Redox Sign. 2013;19(15):1846–54.
Hahn A, Vonck J, Mills DJ, Meier T, Kühlbrandt W. Structure, mechanism, and regulation of the chloroplast ATP synthase. Science. 2018;360(6398):4318.
McLachlin DT, Bestard JA, Dunn SD. The b and d subunits of the escherichia coli ATP synthase interact via residues in their C-terminal regions. J Biol Chem. 1998;273(24):15162–8.
Berry JO, Yerramsetty P, Zielinski AM, Mure CM. Photosynthetic gene expression in higher plants. Photosynth Res. 2013;117(1–3):91–120.
Valegard K, Hasse D, Andersson I, Gunn LH. Structure of rubisco from Arabidopsis thaliana in complex with 2-carboxyarabinitol-1, 5-bisphosphate. Acta Crystallogr. 2018;74(Pt 1):1–9.
Wilson RH, Hayer-Hartl M. Complex Chaperone Dependence of Rubisco Biogenesis. Biochemistry. 2018;57(23):3210-6.
Joshi J, Mueller-Cajar O, Tsai YC, Hartl FU, Hayerhartl M. Role of small subunit in mediating assembly of red-type form I rubisco. J Biol Chem. 2015;290(2):1066–74.
Krapp A, Hofmann B, Schafer C, Stitt M. Regulation of the expression of rbcS and other photosynthetic genes by carbohydrates: a mechanism for the ‘sink regulation’ of photosynthesis? Plant J. 1993;3:817–28.
Cheng SH, Moore B, Seemann JR. Effects of short-and long-term elevated CO2 on the expression of Ribulose-1,5-bisphosphate carboxylase/oxygenase genes and carbohydrate accumulation in leaves of Arabidopsis thaliana (L.) Heynh. Plant Physiol. 1998;116(2):715–23.
Bracher A, Sharma A, Starling-Windhof A, Hart FU, Hayer-Hart M. Degradation of potent rubisco inhibitor by selective sugar phosphatase. Nat Plants. 2015;1(1):14002.
Wang ZY, Portis AR. Dissociation of ribulose-1,5-bisphosphate bound to ribulose-1,5-bisphosphate carboxylase/oxygenase and its enhancement by ribulose-1,5-bisphosphate carboxylase/oxygenase activase-mediatedhydrolysis of ATP. Plant Physiol. 1992;99(4):1348–53.
Galmés J, Aranjuelo I, Medrano H, Flexas J. Variation in rubisco content and activity under variable climatic factors. Photosynth Res. 2013;117(1–3):73–90.
Fukayama H, Mizumoto A, Ueguchi C, Katsunuma J, Morita R, Sasayama D, Hatanaka T, Azuma T. Expression level of rubisco activase negatively correlates with rubisco content in transgenic rice. Photosynth Res. 2018:1–10.
Yamori W, Masumoto C, Fukayama H, Makino A. Rubisco activase is a key regulator of non-steady-state photosynthesis at any leaf temperature and, to a lesser extent, of steady-state photosynthesis at high temperature. Plant J. 2012;71(6):871–80.
Wada S, Suzuki Y, Takagi D, Miyake C, Makino A. Effects of genetic manipulation of the activity of photorespiration on the redox state of photosystem I and its robustness against excess light stress under CO2-limited conditions in rice. Photosynth Res. 2018:1–11.
Maxwell K, Johnson GN. Chlorophyll fluorescence-a practical guide. J Exp Bot. 2000;51(345):659–68.
Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150(1):76–85.
Wu X, Xiong E, Wang W, Scali M, Cresti M. Universal sample preparation method integrating trichloroacetic acid/acetone precipitation with phenol extraction for crop proteomic analysis. Nat Protoc. 2014;9(2):362–74.
Joseph M. Proteomic characterisation of endoplasmic reticulum-derived protein bodies in tobacco leaves. BMC Plant Biol. 2012;12(1):36.
Acknowledgements
We thank Mujitaba Dawuda for the English revision of this manuscript.
Funding
This research was financially supported by the Discipline Construction Funds Project of Gansu Agricultural University, China, (GAU-XKJS-2018-226) and the Science and Technology Major Project of Gansu Province (18ZD2NA006).
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and additional files.
Author information
Authors and Affiliations
Contributions
BHC and JM designed the experiments. XZ and YW conducted the experiments. WFL and XZ analyzed the data. ZHM, SJY and QZ performed the research. WFL and XZ wrote the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Effect of eCO2 on fresh weight, dry weight, leaf area and plant height. (DOC 29 kb)
Additional file 2:
Figure S1. The protein sample analysis by SDS-PAGE. (TIF 508 kb)
Additional file 3:
Table S2. The category with the most DEGs of Cs and C0 compare with CK. (DOC 112 kb)
Additional file 4:
Table S3. The category with the most DEPs of Cs and C0 compare with CK. (DOC 100 kb)
Additional file 5:
Table S4. The top KEGG pathways of Cs versus CK, C0 versus CK and Cs versus C0. (DOC 90 kb)
Additional file 6:
Table S5. Sequences of primer employed in qRT-PCR analysis. (DOC 59 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhao, X., Li, WF., Wang, Y. et al. Elevated CO2 concentration promotes photosynthesis of grape (Vitis vinifera L. cv. ‘Pinot noir’) plantlet in vitro by regulating RbcS and Rca revealed by proteomic and transcriptomic profiles. BMC Plant Biol 19, 42 (2019). https://doi.org/10.1186/s12870-019-1644-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-019-1644-y