
Abstract
Background
Sugar apple (Annona squamosa L.), a popular fruit with high medicinal and nutritional properties, is widely cultivated in tropical South Asia and America. The malformed flower is a major cause for a reduction in production of sugar apple. However, little information is available on the differences between normal and malformed flowers of sugar apple.
Results
To gain a comprehensive perspective on the differences between normal and malformed flowers of sugar apple, cDNA libraries from normal and malformation flowers were prepared independently for Illumina sequencing. The data generated a total of 70,189,896 reads that were integrated and assembled into 55,097 unigenes with a mean length of 783 bp. A large number of differentially expressed genes (DEGs) were identified. Among these DEGs, 701 flower development-associated transcript factor encoding genes were included. Furthermore, a large number of flowering- and hormone-related DEGs were also identified, and most of these genes were down-regulated expressed in the malformation flowers. The expression levels of 15 selected genes were validated using quantitative-PCR. The contents of several endogenous hormones were measured. The malformed flowers displayed lower endogenous hormone levels compared to the normal flowers.
Conclusions
The expression data as well as hormone levels in our study will serve as a comprehensive resource for investigating the regulation mechanism involved in floral organ development in sugar apple.
Similar content being viewed by others


Background
Sugar apple (Annona squamosa L.), a member of the Annonaceae family, is an important medicinal plant that is widely distributed in tropical South Asia and America [1]. In addition to its pharmaceutical value, sugar apple is used in juices, jellies and compotes [2]. Due to its medicinal and nutritional properties, sugar apple is a popular tropical fruit in China [3]. The flowering of sugar apple is a complex process that is regulated by various environmental factors, such as improper shoot state, low temperature or short day conditions [4]. Malformed flowers are the major reason for the low percentage of fertile fruit in cultivated sugar apple. However, the available information on the floral organ development-related genes in the genus Annona is very scarce.
Flowering is a vital developmental process in the life cycles of plants [5]. The coordinated transition from vegetative growth to reproductive development is required for successful reproduction in higher plants [6]. In the model plant Arabidopsis thaliana, the specification of floral organ identities is regulated by a large number of floral genes, most of which encode transcription factors. Based on the phenotypes of Arabidopsis mutants, a classical ABC model was developed [7]. According to the model, most of the floral organ identity-related genes can be grouped into three gene classes: A, B and C. In detail, sepals are identified by Class A genes; petals are identified by Class A together with Class B genes; stamens are identified by Class B genes together with Class C genes; and carpels are identified by Class C genes [8]. Recently, the ABC model was extended and modified by genes (Class E) that are essential for the specification of all types of floral organs [9]. The functions of homeotic genes are highly conserved across different species, allowing us to analyze and identify potential genes that are related to flower development in sugar apple.
Various environmental and endogenous signals are associated with an array of biochemical and cellular processes during the formation of floral organs [10]. Among the signals, phytohormones are endogenously occurring compounds involved in the floral transition, flowering and floral organ development [11]. Several classical phytohormones, such as gibberellins (GAs), auxins, cytokinins (CKs), ethylene, abscisic acid (ABA), jasmonates (JAs), salicylic acid (SA) and brassinosteroids (BRs), have been implicated in the flowering-time pathway [11]. GAs are a class of phytohormones that function not only to induce the transition to flowering, but also to control flowering-time [12]. The role of GAs in the floral transition reflects their recognized function as integrators through DELLA-mediated pathways [13]. GAs promote flowering by triggering the expression of a series of floral integrator encoding genes, including SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1, LEAFY and FLOWERING LOCUS T, (in the inflorescence and floral meristems [14, 15]. Auxins, another classical hormone with a floral-inductive signalling role, were reported as regulators of embryonic and postembryonic development. There is a close correlation between the endogenous auxin content and the flowering time [16]. Several proteins, such as TRANSLOCATED PROMOTER REGION, SUPPRESSOR OF AUXIN RESISTANCE3 and HASTY, are involved in both auxin responses and flowering-time control [17]. CKs, together with auxins, regulate both the division cycle and meristem homeostasis, and may promote the floral transition [18]. In Arabidopsis, applications of exogenous CKs accelerate flowering, though the molecular mechanism underlying this is largely unknown [19].
Thus, there is limited data on the molecular basis of floral initiation and differentiation in the genus Annona. Without reference genomes, de novo sequencing is an effective approach for the identification of candidate genes in most trees, including melon, litchi and Chinese cherry [20,21,22]. In our study, two independent cDNA libraries, one of a normal flower and one of a malformed sugar apple flower, were constructed for Illumina RNA sequencing (RNA-seq). The annotation of the transcriptome sequences allowed for the identification and analysis of potential genes related to floral organ development in sugar apple.
Methods
Plant material and sampling
In our study, 10-year-old adult trees of A. squamosa cv. ‘Bendi’ were cultivated in a 4 × 4 m arrangement with drip irrigation and fertilizer applications as required. The location of the trees was the Ling Nan Normal University field experimental station in Zhanjiang City (Guangdong Province, China). The flowers before open were collected from a normal flower and a malformed flower. All of the flower samples were frozen immediately in liquid nitrogen, and stored at −80 °C for further studies.
cDNA library preparation and Illumina sequencing for transcriptome analysis
Total RNA was extracted using a TRIzol Kit according to the manufacturer’s protocol (Promega, Beijing, China). Residual DNA contamination was removed by RNase-free DNase I (TaKaRa, Dalian, China). RNA quality was verified by RNase-free agarose gel electrophoresis, and the total RNA concentration was measured using a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) at 260 nm and 280 nm. RNA samples with 260 nm/280 nm ratios between 1.8 and 2.0 were used for subsequent analyses. Each cDNA library was constructed by mixing three independent replicate samples. The library of the normal flower was named ‘NF’, while the library of the malformed flower was named ‘MF’. The two libraries were used for the comparative analysis of transcriptome sequencing using the Illumina HiSeq™ 2500 platform by Gene Denovo Co. (Gene Denovo, Guangzhou, China). Raw reads were generated in a paired-end format, and the NF and MF transcriptome data sets were deposited in the GenBank Short Read Archive under accession number SRA508784.
De novo assembly and functional annotation of Illumina sequencing
Low-quality reads (with more than 5% unknown bases) and adaptor sequences were filtered and removed, and then the clean reads were assembled using Trinity software to generate unique consensus contigs [23]. All of the contigs were calculated using a sequence clustering software, and the longest sequences were defined as unigenes. The assembled sugar apple unigenes were aligned to several protein databases, such as NCBI Nr protein (http://www.ncbi.nlm.nih.gov), Swiss-Prot protein (http://www.expasy.ch/sprot), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway (http://www.genome.jp/kegg) and Clusters of Orthologous Groups (COG) (http://www.ncbi.nlm.nih.gov/COG), using the BLASTX algorithm with an E-value <0.00001. For unigene annotation, the Blast2GO program was used to produce the gene ontology (GO) annotation of unigenes, and a BLAST algorithm-based search against the KEGG database (http://www.genome.jp/kegg/) was used to analyze protein products of metabolic processes and related gene functions in cellular processes [24].
Analysis and mapping of digital gene expression (DGE) tags
To map DGE tags, raw data were filtered to remove the low quality tags (sequences with a number of unknown ‘N’s), empty tags (sequences only containing the adaptors), and tags with only one copy number (sequences from sequencing errors). For annotation, cleaned tags containing CATG and the 21-bp tag sequence were mapped to our transcriptome reference database. Initially, the tags that mapped to multiple genes were filtered out. Then, the remaining tags were treated as unambiguous tags for the gene expression analysis. The number of unambiguous tags of each gene was calculated and then normalized to the number of transcripts per million clean tags. The differentially expressed tags between NF and MF were used for further analysis and mapping.
Identification of differentially expressed genes (DEGs)
Reads assoicated with each unigene were mapped to the transcriptome using the alignment software Bowtie 0.12.8 (https://sourceforge.net/projects/bowtie-bio/files/bowtie/0.12.8/). To calculate unigene expression, the number of mapped reads for each unigene was counted and normalized into a reads per kb per million reads (RPKM) value. To determine significant differences, a false discovery rate < 0.001 and an absolute value of log2 ratio > 1 were set as thresholds. DEGs between the two flower samples were calculated using the edgeR package [25]. GO and KEGG enrichment analyses of the DEGs between two flower samples were performed according to a previous reported method [26].
Quantitative real-time PCR (qRT-PCR) validation
Total RNA was extracted from the same sample that were used for sequencing. In total, 3 μg RNA of each sample was used for reverse transcription. The cDNA was synthesized using ReverAid First Strand cDNA Synthesis Kit (Thermo Scientific. Shanghai, China). QRT-PCR was performed using the SYBR Premix Ex Taq Kit (TaKaRa, Dalian, China) and an ABI PRISM 7700 DNA Sequence Detection System (Applied Biosystems, Shanghai, China). Three independent cDNA samples from “NF” and “MF” were used for qRT-PCR validation. The primer sequences were designed using Primer Premier 5 software (Premier Biosoft International, Palo Alto, CA, USA). The sugar apple Actin gene was used as an internal standard to calculate relative fold-differences based on comparative cycle threshold (2−ΔΔCt) values. Then, ddH2O was used as no-template control. The qRT-PCR procedure was as follows: 1 μL of a 1/10 dilution of cDNA in H2O was added to 5 μL of 2× SYBR® Green buffer, with 0.1 μM of each primer and H2O to a final volume of 10 μL. The reactions were run as follows: 50 °C for 2 min and 95 °C for 10 min, followed by 40 cycles of 95 °C for 30 s, 56 °C for 30 s and 72 °C for 30 s in 96-well optical reaction plates [27]. Each real-time PCR reaction was performed three times.
Measurements of various hormones
For the exogenous hormone contents analysis, independent samples from normal and malformed sugar apple flowers were harvested, immediately frozen in liquid nitrogen and stored at −80 °C for further extractions. Exogenous indole-3-acetic acid (IAA) contents were determined using a FOCUS GC-DSQII (Thermo Fisher Scientific Inc., Austin, TX, USA) as described previously [28]. Endogenous GA was measured using the nano-LC-ESI-Q-TOF-MS analysis method described previously [29]. In additionally, ABA and zeatin ribosides (ZRs) were detected using a UFLC-MS/MS system described by Kasote et al. [30].
Statistical analysis
Significant differences between different samples were calculated using a one-way analysis of variance with a Tukey’s test (at a significance level of α = 0.01) in Excel software. All of the expression analyses were performed for three biological replicates. All reported values represent the averages of three replicates, and data are expressed as the mean plus or minus the standard deviation (mean ± SD).
Results
Sequencing, de novo assembly and sequence annotation
To obtain a reference transcriptome for the A. squamosa flower, two independent RNA-seq libraries were constructed using RNA samples from normal flowers and malformed flowers, separately. The phenotypic characterizations of normal and malformed flowers is shown in Fig. 1a. In detail, the differentiation and development of petals are irregular in the malformed flowers. Compared with normal flowers at the same stage, the termination of the ovule development occurs before blastophore formation in malformed flowers. Additionally, no female gametes were observed in malformed flowers.

Illumina sequencing of sugar apple flowers. a The phenotypes of normal and malformation flowers in sugar apple. b Classification of raw reads generated by Illumina sequencing. c The length distribution of assembled transcripts in sugar apple. d The length distribution of assembled unigenes in sugar apple
In our study, a large number of raw reads, including 98.15% of clean reads, 0.88% of only adaptors and 0.97% of N-containing reads, were obtained (Fig. 1b). A total of 70,189,896 reads were integrated and assembled into 55,097 unigenes with a mean length of 783 bp. The detailed information of the obtained reads is listed in Additional file 1. The size distributions of clean reads and unigenes in sugar apple are shown in Fig. 1c and d. For clean reads, 3% of the reads were >3000 bp in length and a majority of the reads (58%) were <500 bp in length. Only 1% of the unigenes were >10,000 bp in length, and the majority of unigenes were between 11 bp and 50 bp.
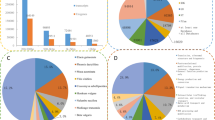
For the annotation, the unigenes were queried against several databases, including Nr, SwissProt, KEGG and COG, using BLASTX algorithm-based software. In total, 55,097 unigenes could be identified in the Nr database, 24,308 unigenes displayed a significant similarity to known proteins in the SwissProt database, and 8805 unigenes were annotated in both the COG and KEGG databases based on sequence homologies (Fig. 2a). Approximately 8784 unigenes were matched to a homolog in all four databases. The distribution of the E-values of the annotated unigenes is shown in Fig. 2b.

Annotation of assembled sugar apple unigenes. (a) The number of unigenes annotated by different databases, including Nr, Swissprot, COG and KEGG, were showed in a Venn diagram. (b) Distribution of E value in various databases. (c) COG classification of all unigenes of sugar apple. Proportion of each second level COG term belonged to “Signal transduction mechanisms” and “Cell cycle control, cell division”
For the COG classification, 13,548 unigenes were grouped into 25 functional classifications. The largest term, “General function prediction only”, contained 2415 unigenes. “Replication, recombination and repair”, “Cell wall/membrane/envelope”, “Posttranslational modification” and “Signal transduction mechanisms” shared a large percentage of unigenes. In addition, only nine unigenes were classified into “Cell motility”, and two unigenes were classified into “Nuclear structure”. Furthermore, two flower development-related COG terms, “signaling transduction mechanisms” and “Cell cycle control, cell division”, were analyzed in detail (Fig. 2c).
GO and KEGG classifications of unigenes
In sugar apple, most of the unigenes could be assigned to 46 functional terms that belonged to three GO categories, biological process, cellular component and molecular function. For biological process, “metabolic processes” (6128 unigenes) and “cellular processes” (5721 unigenes) were dominant terms; for cellular component, the dominant terms were “cell” (6377 unigenes), “cell part” (6227 unigenes) and “organelle” (6128 unigenes); and for molecular function, a large percentage of unigenes were related to “metabolic process” (6707 unigenes), “catalytic activity” (5942 unigenes) and “binding” (5168 unigenes) (Additional file 2).
Furthermore, all of the unigenes mapped to canonical pathways in the KEGG database. Our data showed that 7191 unigenes from sugar apple were assigned to 124 KEGG pathways (Additional file 3). The most represented KEGG pathway was “metabolic pathways” with 1940 unigenes (ko01100). Additionally, 959 unigenes were associated with the KEGG pathway “biosynthesis of secondary metabolites”, 335 unigenes were mapped to “ribosome”, and 219 unigenes were assigned to “protein processing in endoplasmic reticulum”.
Screening and classification of DEGs between normal and malformed flowers
To compare the DEGs between normal and malformed flowers, RPKM values were determined to calculate the read density for each unigene. Expression profiles of the DEGs between normal and malformed flowers in sugar apple are shown in a heatmap (Fig. 3a). A total of 12,664 DEGs were identified, including 1690 up-regulated genes and 10,974 down-regulated genes (Fig. 3b). A significance analysis of the DEGs between normal and malformed flowers was visualized using a volcano plot (Fig. 3c). An analysis of the biological functions of these DEGs was performed. Among these DEGs, 2822 unigenes could be annotated from GO-based sequence homologies. For the GO classification, the top five largest GO terms in biological process were “metabolic process”, “cellular process”, “single-organism process”, “response to stimulus” and “localization”; in cellular component, the top five largest GO terms were “cell”, “cell part”, “organelle”, “organelle part” and “macromolecular complex”; and in molecular function, “metabolic process”, “catalytic activity”, “binding”, “single-organism process” and “cellular process” were the five largest GO terms (Fig. 3d).

Transcriptional variation normal and malformed flowers in sugar apple. a Expression profiles of the differential expressed genes between normal and malformed flowers in sugar apple were showed by a heatmap. b The numbers of up-regulated genes and down-regulated genes in malformed flower compared to normal flower. c Significance analysis of all DEGs between normal and malformed flowers by a volcanoplot. d GO enrichment analysis of DEGs between normal and malformed flowers
Identification of the floral organ development-associated TF-encoding genes and MADS-box genes
In total, 701 TF genes were identified as DEGs (Additional file 4). The top five largest differential expressed TF families were: ERF (53 members), bHLH (42 members), MYB-related (38 members), FAR1 (37 members) and NAC (36 members). Moreover, several hormone-related TFs also were identified as DEGs, including 24GRASs and 9 ARFs. In our study, 21 MADS-box genes were revealed from sugar apple flowers (Additional file 5). Among these MADS-box genes, four genes, such as AGL9, SEP1, AP1-like, were identified in MIKC family and 17 genes, such as AP3-like, AGL63 and PISTILLATA-like.
Identification of key flowering- and flower development-related DEGs
A large number of flowering- and flower development-related genes play important roles in the entire flower developmental cycle [31]. In total, 18 DEGs in sugar apple showed homology to known flowering- and flower development-related genes in the NCBI and UniProt databases (Additional file 6). For example, two flowering promoting factor genes (Unigene0041725 and Unigene0054870) showed higher expression levels in the malformed flowers than in the normal flowers. Additionally, four early flowering protein genes (Unigene0023978, Unigene0023976, Unigene0033968 and Unigene0008711), one flowering locus T gene (Unigene0000043), one flowering time control protein FY-like gene (Unigene0025538), three flowering time control protein FPA-like genes (Unigene0005475, Unigene0019814 and Unigene0005474), and one flowering time control protein FCA-like gene (Unigene0024514), were also identified as DEGs.
Based on their GO terms (GO:0009908), 41 DEGs were identified as flower development-related genes (Additional file 7). Interestingly, two genes, a probable histone H2A variant 3-like gene (Unigene0017118) and ODORANT1-like gene (Unigene0047135), were largely induced (> 5 fold) in the malformed flowers. Most of other identified flower development-related DEGs were reduced in the malformed flowers.
Identification of hormone-related DEGs
In our study, a large number of hormone-related DEGs were identified between normal and malformed flowers. The KEGG analysis assigned most of the DEGs to key components involved in various hormonal signaling pathways (Fig. 4a). Interestingly, a large number of hormone-related DEGs belonged to auxin and CK signaling pathways. An overview of the various hormonal signaling networks in sugar apple is shown in Fig. 4b. Most hormone-related DEGs were predominantly expressed in normal flowers.

Identification and differential analysis of the hormonal network between normal and malformed flowers in sugar apple. a The number of the DEGs encoding the key components involved in various hormonal signaling pathways. b Overview of various hormonal signaling network in sugar apple. Red indicated the normal flower predominantly expressed genes and green indicated the malformed flower predominantly expressed genes
Firstly, DEGs related to auxin were analyzed. For auxin biosynthesis and metabolism, 5 YUCCA genes and 13 GH3 genes were identified as DEGs; for auxin transport, 14 efflux carrier-encoding genes and seven influx carrier-encoding genes were identified as DEGs. For the downstream response, 35 auxin-induced genes, 3 auxin-repressed genes, 39 auxin response factor-encoding genes, and 21 auxin/IAA-encoding genes were identified as DEGs. Secondly, DEGs associated with ABA were identified. For ABA biosynthesis, seven CYP707A1 genes were obtained. For ABA receptors and transporters, seven ABA1-encoding genes and four PYL-encoding genes were identified. For ABA downstream responses, 21 DEGs were annotated as FTA, KEG, AIP, PED and HVA genes. Then, DEGs related to GA were analyzed. For GA biosynthesis, 23 DEGs encoding GA20OX1, GA3OX1 and GA2OX2 were identified, and 3 DEGs were annotated as GA receptors. For GA downstream responses, eight DEGs were identified as GA responsive genes and eight DEGs were identified as GRAS. Furthermore, 53 DEGs were mapped to the CK pathway. In total, 13 DEGs were associated with CK biosynthesis and metabolism (such as CYP and CKX), 10 DEGs were identified as HK/CRE-encoding genes, and 31 CK downstream responsive genes were identified (Fig. 5a).

Transcript abundance changes of hormone-related genes. a The detailed information on genes involved different hormone signaling pathway. The key components in red cycle indicated genes related to hormone biosynthesis and metabolism; the key components in green cycle indicated genes encoding receptor and transporter; and the key components in blue cycle indicated genes related to downstream response. b Expression changes of the genes associated with different hormones, including auxin, ABA, GA and cytokinin. Red indicates up-regulated genes and blue indicates down-regulated genes
Based on the RPKM values, the expression levels of the hormone-related unigenes were analyzed and shown in Fig. 5b. The expression levels of most hormone-related unigenes in the normal flowers were greater than those in the malformed flowers.
Validation of the expression of several key hormone-related genes
To verify the differential expression levels of some key hormone-related genes identified by RNA-seq, a qRT-PCR assay with independent samples collected from normal and malformed flowers was performed. In total, 15 key flower hormone-related genes and eight floral organ development-associated TF-encoding genes were randomly selected to validate the RNA-seq data. The expression levels of these selected genes were basically consistent with RNA-seq results (Fig. 6). Furthermore, the expression of these selected genes were analyzed with independent samples collected from the flowers at different developmental stages [32]. Expression profiles of these selected genes during the flower development process were showed in Additional files 8 and 9 . The primer sequences are listed in Additional file 10.

Real-time quantitative PCR validation of several selected hormone-related genes. Total RNA was extracted from normal and malformation flowers. The histogram shows the relative expression level of these genes with respect to the ACTIN in hickory. The specific identities of the genes: Unigene0001697 (auxin-induced protein 5NG4-like), Unigene0004014 (auxin response factor 5-like), Unigene0037190 (IAA29-like), Unigene0033914 (PIN5), Unigene0004847 (gibberellin-regulated protein 9), Unigene0054219 (GRAS family transcription factor), Unigene0010774 (gibberellin 2-oxidase 2), Unigene0048844 (gibberellin 3-beta-hydroxylase), Unigene0011098 (APRR2), Unigene0018960 (PRR95), Unigene0010916 (cytokinin dehydrogenase 3-like), Unigene0051789 (3-ketoacyl-CoA thiolase 2), Unigene0023736 (zeaxanthin epoxidase), Unigene0006250 (ABA 8′-hydroxylase), Unigene0033398 (PYR1-like). The data were analyzed by three independent repeats, and standard deviations were shown with error bars. Significant differences in expression level were indicated by “*”
Endogenous hormone measurements
To examine the changes in endogenous hormones between normal and malformed flowers, the IAA, ABA, GA and ZRs contents were measured. Three independent samples collected from different flowers were used for endogenous hormone measurements. The IAA, ABA, GA and ZRs contents were reduced in malformed flowers (Fig. 7). In detail, compared with normal flowers, the IAA contents decreased from 321.23 to 185.25 nmol·g−1, the ABA contents decreased from 35.63 to 12.32 nmol·g−1, the GA contents decreased from 112.36 to 87.23 nmol·g−1, and the ZRs contents decreased from 265.32 to 145.68 nmol·g−1 in malformed flowers.

Endogenous hormone measurements. The contents of endogenous hormones, including IAA, ABA, GA and ZRs, between normal and malformation flowers were measured. Three independent samples collected from different flowers were used for endogenous hormone measurements. Significant differences in contents were indicated by “*”
Discussion
Sugar apple is a popular fruit tree with a high commercial value in subtropical and tropical areas [1]. Without a public genome database, detailed information on the genes involved in their growth and development is unavailable for sugar apple studies. A DEG analysis is a good tool for studying the temporal regulation of gene expression [33]. Our previous work constructed independent cDNA libraries of four different flower stages in A. squamosa for Illumina RNA-seq. A large number of flower developmental stage-related genes have been identified [32]. In the present study, transcriptome data from normal and malformed flowers at the same developmental stage (the mature flowers with partially opened petals) were used to uncover the differences between normal and malformed flowers and provide a more adequate resource to study sugar apple.
In model plants, various TF families, such as MYB, MADS-box, NAC, ARF and bHLH, have been reported to be involved in floral development [34,35,36,37,38]. Therefore, the differential expression levels of TF genes between normal and malformed flowers were analyzed in our study. Our previous study identified a large number of flower development-related TF families, including bHLH, NAC, B3, MYB-related and bZIP. MYB factors promote petal and stamen growth in Arabidopsis. For example, MYB21 fed back negatively on the expression of JA biosynthesis-related genes to control flower development by decreasing the JA level [39]. Our previous study identified a large MYB-related TFs, suggesting an important role for MYB factors in the floral organ development of sugar apple [32]. In the present study, the MYB family contained the largest number of differentially expressed TFs (59 members). Additionally, bHLH is another large TF family that regulates various flower developmentally related processes [40]. In sugar apple, the bHLH family had the second largest number of differentially expressed TFs (35 members) between normal and malformed flowers. Our previous study showed that bHLH is the largest TF family involved in flower development of sugar apple, indicating a involvement of MYB and bHLH families in the differences between normal and malformed flowers.
The Arabidopsis TFs ARF6 and ARF8 are expressed in multiple flower tissues, such as sepals, petals and stamen filaments [41]. Moreover, these two genes function in different organs to promote the transition from closed buds to mature fertile flowers [42]. ARF1 and ARF2 regulate senescence and floral organ abscission in Arabidopsis [43]. In sugar apple, nine ARFs were identified as differentially expressed TFs between normal and malformed flowers, indicating that ARF-mediated auxin signaling participated in flower development.
It has been reported that morphogenesis of floral organs was under control of several MADS-box genes. Loss-of-function of any essential MADS-box genes may result in homeotic conversion of floral organs [44]. For example, four orchid AGL9-like MADS-box genes play roles in floral transition and formation in Arabidopsis [45]. In the present study, four MIKC-type MADS-box genes were identified, including an AGL9 gene, which may involved in floral transition of sugar apple. Besides, SEPALLATA subfamily MADS-box protein was reported to positively control spikelet meristem identify in rice [46]. A SEPALLATA 1-like protein encoding gene also was identified in sugar apple. The data provides a more comprehensive information of the floral organ development process in sugar apple.
Based on the DEGs analysis, a large number of auxin-related genes, including ARFs, Aux/IAAs, and GHs, were identified as DEGs. The phytohormone auxin is involved in regulating many aspects of plant growth and development [47, 48]. A classical role of auxin in the formation of flowers at the periphery of the reproductive shoot apex was revealed by genetic evidence [49]. For example, mutations in PIN1 or PID, auxin efflux carrier-encoding genes, lack flowers [50]. At the early stage, the development of female gametophytes is controlled by the AtPIN1-mediated auxin flux [51]. In our study, the identified auxin influx carrier-encoding genes and most of the identified auxin efflux carrier-encoding genes were down-regulated in the malformed flowers. In Arabidopsis, ARF4 is associated with flower patterning [52]. Another ARF gene, AtARF3, integrates the functions of AGAMOUS and APETALA2 in floral meristem determinacy [53, 54]. Here, the expression of most of the identified ARFs and Aux/IAAs was also reduced in the malformed flowers. Thus, auxin and auxin transport may be required for floral meristem determinacy and flower patterning in sugar apple.
CK plays pivotal roles in many aspects of plant development, including the formation of male and female functions [55]. In Arabidopsis, the floral homeotic gene APETALA1 directly reduces the biosynthesis of endogenous CK by promoting the expression of the CK degradation gene CYTOKININ OXIDASE/DEHYDROGENASE3 and inhibiting the expression of the CK biosynthetic gene LONELY GUY1 [56]. In our study, a number of CK biosynthesis-related genes, including 3 CYPs and 10 CKXs, were identified as DEGs in sugar apple. Decreases in the biosynthesis of CK may cause the malformed flower phenotype.
In additional, GA and ABA play important roles in flower development. For example, effects of exogenous GA3 applications on grape flowers have been analyzed. An RNA-seq transcriptome analysis suggested that the morphology of grape inflorescences may be controlled by the biosynthesis and signaling of GA3 [57]. In Arabidopsis, a GA-deficient mutant, ga1–3, displayed the retarded growth of four whorls in the floral organs, and its flower phenotypes could be rescued by the application of exogenous GA [58]. GA-INSENSITIVE DWARF1 (GID1), a soluble protein with a high similarity to hormone-sensitive lipases, was first cloned in rice as a GA receptor [59]. In our study, two GID-encoding genes were identified as DEGs, suggesting an involvement of GA signaling in sugar apple’s flower development. For GA biosynthesis, the GA2ox family genes were identified in various plant species [60]. Based on our transcriptome data, some GA3OX1 family genes, such as Unigene0016441, were largely reduced in malformed flowers, while several GA2OX2 family genes, such as Unigene0010774 and Unigene0004102, were induced in malformed flowers. This suggested that a diversity of regulatory mechanisms are involved in GA biosynthesis during flower development. A number of transcriptomes revealed that there is a close relationship between ABA signaling and flower development in various plant species, such as Gerbera hybrida [61]. Our data confirmed that ABA-related genes also showed significant expression changes between normal and malformed flowers.
Furthermore, malformed flowers displayed lower endogenous hormone levels compared with the normal flowers. This was in agreement with the decreases in the expression levels of hormone-related genes in malformed flowers. In Arabidopsis, increased auxin levels attribute the initiation of flower formation in the shoot apex [62]. Addition to auxin, cytokinin accumulation also causes alteration of flower development in Arabidopsis [63]. In sugar apple, low auxin and cytokinin levels may affect downstream processes of flower development and lead to malformed flowers. ABA alters the number of carpels in Arabidopsis [64]. Compared with normal flowers, lower ABA level may affect the formation of carpels in malformed flowers. Recent publications have established that GA is involved in stamen development. For example, mutations in GA receptors affect the elongation of stamens [65]. GA concentration may play a role in stamen development in sugar apple.
In our previous work, genes associated with floral transition and flower development were identified in sugar apple [32]. In the present study, most selected DEGs showed significantly expression changes during the flower development process, suggesting a causal relationship to the defects associated with the malformed flowers.
Conclusions
In summary, two independent cDNA libraries from normal and malformed flowers of sugar apple were separately constructed and sequenced. A large number of DEGs were identified between normal and malformed flowers. The expression changes of hormone-related unigenes were analyzed and validated using a qRT-PCR analysis. The expression analysis, together with the hormone contents, provides a comprehensive resource for investigating the differential expressed genes between normal and malformed flowers.
Abbreviations
- ABA:
-
abscisic acid
- BRs:
-
brassinosteroids
- CKs:
-
cytokinins
- COG:
-
Clusters of Orthologous Groups
- DEGs:
-
differentially expressed genes
- DGE:
-
digital gene expression
- GAs:
-
gibberellins
- GO:
-
gene ontology
- IAA:
-
indole-3-acetic acid
- JAs:
-
jasmonates
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- qRT-PCR:
-
quantitative real-time PCR
- RNA-seq:
-
RNA sequencing
- RPKM:
-
reads per kb per million reads
- SA:
-
salicylic acid
- ZRs:
-
zeatin ribosides
References
Hollerhage M, Rosler TW, Berjas M, Luo R, Tran K, Richards KM, et al. Neurotoxicity of dietary supplements from Annonaceae species. Int J Toxicol. 2015;34(6):543–50.
Deng GF, Xu DP, Li S, Li HB. Optimization of ultrasound-assisted extraction of natural antioxidants from sugar apple (Annona squamosa L.) peel using response surface methodology. Molecules. 2015;20(11):20448–59.
Liu K, Li H, Yuan C, Huang Y, Chen Y, Liu J. Identification of phenological growth stages of sugar apple (Annona squamosa L.) using the extended BBCH-scale. Sci Hortic. 2015;181:76–80.
Olesen T, Muldoon SJ. Effects of defoliation on flower development in atemoya custard apple (Annona cherimola mill.×a. squamosa L.) and implications for flower-development modelling. Aust J Bot. 2012;60(2):160–4.
Hong Y, Jackson S. Floral induction and flower formation--the role and potential applications of miRNAs. Plant Biotechnol J. 2015;13(3):282–92.
Li C, Gu M, Shi N, Zhang H, Yang X, Osman T, et al. Mobile FT mRNA contributes to the systemic florigen signalling in floral induction. Sci Rep. 2011;1:73.
Mendoza L, Thieffry D, Alvarez-Buylla ER. Genetic control of flower morphogenesis in Arabidopsis thaliana: a logical analysis. Bioinformatics. 1999;15(7–8):593–606.
Causier B, Schwarz-Sommer Z, Davies B. Floral organ identity: 20 years of ABCs. Semin Cell Dev Biol. 2010;21(1):73–9.
Theissen G. Development of floral organ identity: stories from the MADS house. Curr Opin Plant Biol. 2001;4(1):75–85.
Sablowski R. Flowering and determinacy in Arabidopsis. J Exp Bot. 2007;58(5):899–907.
Davis SJ. Integrating hormones into the floral-transition pathway of Arabidopsis thaliana. Plant Cell Environ. 2009;32(9):1201–10.
Pang J, Wang L, Jiangqin HU, Xiang T, Liang H. Synergistic promotion of gibberellin and cytokinin on direct regeneration of floral buds from in vitro cultures of sepal segments in sinningia speciosa hiern. In Vitr Cell Dev Biol - Plant. 2006;42(5):450–4.
Davière JM, Lucas MD, Prat S. Transcriptional factor interaction: a central step in DELLA function. Curr Opin Genet Dev. 2008;18(4):295–303.
Liu C, Chen H, Er HL, Soo HM, Kumar PP, Han JH, et al. Direct interaction of AGL24 and SOC1 integrates flowering signals in Arabidopsis. Development. 2008;135(8):1481–91.
Zhang MZ, Ye D, Wang LL, Pang JL, Zhang YH, Zheng K, et al. Overexpression of the cucumber LEAFY homolog CFL and hormone treatments alter flower development in gloxinia (Sinningia speciosa). Plant Mol Biol. 2008;67(4):419–27.
Thingnaes E, Torre S, Ernstsen A, Moe R. Day and night temperature responses in Arabidopsis: effects on gibberellin and auxin content, cell size, morphology and flowering time. Ann Bot. 2003;92(4):601–12.
Jacob Y, Mongkolsiriwatana C, Veley KM, Kim SY, Michaels SD. The nuclear pore protein AtTPR is required for RNA homeostasis, flowering time, and auxin signaling. Plant Physiol. 2007;144(3):1383–90.
Zhao Y. The role of local biosynthesis of auxin and cytokinin in plant development. Curr Opin Plant Biol. 2008;11(1):16–22.
Bernier G, Perilleux C. A physiological overview of the genetics of flowering time control. Plant Biotechnol J. 2005;3(1):3–16.
Corbacho J, Romojaro F, Pech JC, Latche A, Gomez-Jimenez MC. Transcriptomic events involved in melon mature-fruit abscission comprise the sequential induction of cell-wall degrading genes coupled to a stimulation of endo and exocytosis. PLoS One. 2013;8(3):e58363.
Lu X, Kim H, Zhong S, Chen H, Hu Z, Zhou B. De novo transcriptome assembly for rudimentary leaves in litchi chinesis Sonn. And identification of differentially expressed genes in response to reactive oxygen species. BMC Genomics. 2014;15:805.
Zhu Y, Li Y, Xin D, Chen W, Shao X, Wang Y, et al. RNA-Seq-based transcriptome analysis of dormant flower buds of Chinese cherry (Prunus pseudocerasus). Gene. 2015;555(2):362–76.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat Biotechnol. 2011;29(7):644–52.
Shen C, Guo H, Chen H, Shi Y, Meng Y, Lu J, et al. Identification and analysis of genes associated with the synthesis of bioactive constituents in Dendrobium officinale using RNA-Seq. Sci Rep. 2017;7(1):187.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40.
Shen C, Yang Y, Liu K, Zhang L, Guo H, Sun T, et al. Involvement of endogenous salicylic acid in iron-deficiency responses in Arabidopsis. J Exp Bot. 2016;67(14):4179–93.
Shen C, Yue R, Bai Y, Feng R, Sun T, Wang X, et al. Identification and analysis of Medicago truncatula auxin transporter gene families uncover their roles in responses to Sinorhizobium meliloti infection. Plant cell physiol. 2015;56(10):1930–43.
Shen C, Yue R, Sun T, Zhang L, Yang Y, Wang H. OsARF16, a transcription factor regulating auxin redistribution, is required for iron deficiency response in rice (Oryza sativa L.). Plant Sci. 2015;231:148–58.
Chen ML, Fu XM, Liu JQ, Ye TT, Hou SY, Huang YQ, et al. Highly sensitive and quantitative profiling of acidic phytohormones using derivatization approach coupled with nano-LC-ESI-Q-TOF-MS analysis. J Chromatogr B. 2012;905:67–74.
Kasote DM, Ghosh R, Chung JY, Kim J, Bae I, Bae H. Multiple reaction monitoring mode based liquid chromatography-mass spectrometry method for simultaneous quantification of brassinolide and other plant hormones involved in abiotic stresses. Int J anal chem. 2016;2016:7214087.
Sui S, Luo J, Ma J, Zhu Q, Lei X, Li M. Generation and analysis of expressed sequence tags from Chimonanthus praecox (Wintersweet) flowers for discovering stress-responsive and floral development-related genes. Comp funct genomics. 2012;2012:134596.
Liu K, Feng S, Pan Y, Zhong J, Chen Y, Yuan C, et al. Transcriptome analysis and identification of genes associated with floral transition and flower development in sugar apple (Annona squamosa L.). Front Plant Sci. 2016;7:1695.
Hao J, Guo H, Shi X, Wang Y, Wan Q, Song Y et al. Comparative proteomic analyses of two Taxus species (Taxus × media and Taxus mairei ) reveals variations in the metabolisms associated with paclitaxel and other metabolites. Plant and Cell Physiology. 2017. doi:10.1093/pcp/pcx128.
Yu C, Guo H, Zhang Y, Song Y, Pi E, Yu C, Zhang L, et al. Identification of potential genes that contributed to the variation in the taxoid contents between two Taxus species (Taxus media and Taxus mairei). Tree Physiol. 2017; doi: 10.1093/treephys/tpx091.
Ning YQ, Ma ZY, Huang HW, Mo H, Zhao TT, Li L et al. Two novel NAC transcription factors regulate gene expression and flowering time by associating with the histone demethylase JMJ14. Nucleic acids research. 2015;43(3):1469-1484
Liu K, Yuan C, Li H, Lin W, Yang Y, Shen C, et al. Genome-wide identification and characterization of auxin response factor (ARF) family genes related to flower and fruit development in papaya (Carica papaya L.). BMC Genomics. 2015;16(1):901.
Matias-Hernandez L, Aguilar-Jaramillo AE, Cigliano RA, Sanseverino W, Pelaz S. Flowering and trichome development share hormonal and transcription factor regulation. J Exp Bot. 2016;67(5):1209–19.
Becker A, Theissen G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol Phylogenet Evol. 2003;29(3):464–89.
Reeves PH, Ellis CM, Ploense SE, Wu MF, Yadav V, Tholl D, et al. A regulatory network for coordinated flower maturation. PLoS Genet. 2012;8(2):e1002506.
Zhang W, Sun Y, Timofejeva L, Chen C, Grossniklaus U, Ma H. Regulation of Arabidopsis Tapetum development and function by DYSFUNCTIONAL TAPETUM1 (DYT1) encoding a putative bHLH transcription factor. Development. 2006;133(16):3085–95.
Nagpal P, Ellis CM, Weber H, Ploense SE, Barkawi LS, Guilfoyle TJ, et al. Auxin response factors ARF6 and ARF8 promote jasmonic acid production and flower maturation. Development. 2005;132(18):4107–18.
Wu MF, Tian Q, Reed JW. Arabidopsis microRNA167 controls patterns of ARF6 and ARF8 expression, and regulates both female and male reproduction. Development. 2006;133(21):4211–8.
Ellis CM, Nagpal P, Young JC, Hagen G, Guilfoyle TJ, Reed JW. AUXIN RESPONSE FACTOR1 and AUXIN RESPONSE FACTOR2 regulate senescence and floral organ abscission in Arabidopsis thaliana. Development. 2005;132(20):4563–74.
Parenicova L, de Folter S, Kieffer M, Horner DS, Favalli C, Busscher J, et al. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: new openings to the MADS world. Plant Cell. 2003;15(7):1538–51.
Chang YY, Chiu YF, Wu JW, Yang CH. Four orchid (Oncidium Gower Ramsey) AP1/AGL9-like MADS box genes show novel expression patterns and cause different effects on floral transition and formation in Arabidopsis thaliana. Plant cell physiol. 2009;50(8):1425–38.
Kobayashi K, Maekawa M, Miyao A, Hirochika H, Kyozuka J. PANICLE PHYTOMER2 (PAP2), encoding a SEPALLATA subfamily MADS-box protein, positively controls spikelet meristem identity in rice. Plant cell physiol. 2010;51(1):47–57.
Yang X, Liu X, Lv W, Li L, Shi Q, Yang J, et al. Reduced expression of BjRCE1 gene modulated by nuclear-cytoplasmic incompatibility alters auxin response in cytoplasmic male-sterile Brassica juncea. PLoS One. 2012;7(6):e38821.
Liu K, Yue R, Yuan C, Liu J, Zhang L, Sun T, et al. Auxin signaling is involved in iron deficiency-induced photosynthetic inhibition and shoot growth defect in rice (Oryza sativa L.). J Plant Biol. 2015;58(6):391–401.
Yamaguchi N, Wu MF, Winter CM, Berns MC, Nole-Wilson S, Yamaguchi A, et al. A molecular framework for auxin-mediated initiation of flower primordia. Dev Cell. 2013;24(3):271–82.
Przemeck GK, Mattsson J, Hardtke CS, Sung ZR, Berleth T. Studies on the role of the Arabidopsis gene MONOPTEROS in vascular development and plant cell axialization. Planta. 1996;200(2):229–37.
Ceccato L, Masiero S, Sinha Roy D, Bencivenga S, Roig-Villanova I, Ditengou FA, et al. Maternal control of PIN1 is required for female gametophyte development in Arabidopsis. PLoS One. 2013;8(6):e66148.
Hunter C, Willmann MR, Wu G, Yoshikawa M, de la Luz Gutierrez-Nava M, Poethig SR. Trans-acting siRNA-mediated repression of ETTIN and ARF4 regulates heteroblasty in Arabidopsis. Development. 2006;133(15):2973–81.
Wang HZ, Hu B, Chen GP, Shi NN, Zhao Y, Yin QC, et al. Application of Arabidopsis AGAMOUS second intron for the engineered ablation of flower development in transgenic tobacco. Plant Cell Rep. 2008;27(2):251–9.
Xu X, Bian J, Liu S, Song H, Shi N, Tao Y, et al. Flower-specific expression of Arabidopsis PCS1 driven by AGAMOUS second intron in tobacco decreases the fertility of transgenic plants. Mol Breed. 2011;27(3):337–46.
Kinoshita-Tsujimura K, Kakimoto T. Cytokinin receptors in sporophytes are essential for male and female functions in Arabidopsis thaliana. Plant Signal Behav. 2011;6(1):66–71.
Han Y, Zhang C, Yang H, Jiao Y. Cytokinin pathway mediates APETALA1 function in the establishment of determinate floral meristems in Arabidopsis. Proc Natl Acad Sci U S A. 2014;111(18):6840–5.
Cheng C, Jiao C, Singer SD, Gao M, Xu X, Zhou Y, et al. Gibberellin-induced changes in the transcriptome of grapevine (Vitis labrusca x V. vinifera) cv. Kyoho flowers. BMC Genomics. 2015;16:128.
Goto N, Pharis RP. Role of gibberellins in the development of floral organs of the gibberellin-deficient mutant, ga1-1, of Arabidopsis thaliana. Can J Bot. 1999;77(7):944–54.
Ren D, Rao Y, Wu L, Xu Q, Li Z, Yu H, et al. The pleiotropic ABNORMAL FLOWER AND DWARF1 affects plant height, floral development and grain yield in rice. J Integr Plant Biol. 2016;58(6):529–39.
Pearce S, Huttly AK, Prosser IM, Li YD, Vaughan SP, Gallova B, et al. Heterologous expression and transcript analysis of gibberellin biosynthetic genes of grasses reveals novel functionality in the GA3ox family. BMC Plant Biol. 2015;15:130.
Li L, Zhang W, Zhang L, Li N, Peng J, Wang Y, et al. Transcriptomic insights into antagonistic effects of gibberellin and abscisic acid on petal growth in Gerbera hybrida. Front Plant Sci. 2015;6:168.
Taoka K, Yanagimoto Y, Daimon Y, Hibara K, Aida M, Tasaka M. The NAC domain mediates functional specificity of CUP-SHAPED COTYLEDON proteins. Plant J. 2004;40(4):462–73.
Li XG, Su YH, Zhao XY, Li W, Gao XQ, Zhang XS. Cytokinin overproduction-caused alteration of flower development is partially mediated by CUC2 and CUC3 in Arabidopsis. Gene. 2010;450(1–2):109–20.
Fitzpatrick AH, Shrestha N, Bhandari J, Crowell DN. Roles for farnesol and ABA in Arabidopsis flower development. Plant Signal Behav. 2011;6(8):1189–91.
Iuchi S, Suzuki H, Kim YC, Iuchi A, Kuromori T, Ueguchi-Tanaka M, et al. Multiple loss-of-function of Arabidopsis gibberellin receptor AtGID1s completely shuts down a gibberellin signal. Plant J. 2007;50(6):958–66.
Acknowledgements
We are grateful to Qian Liu and Yang Liu (from Guangzhou Genedenovo Biotechnology Co., Ltd) for technical support.
Funding
This work was supported by the National Natural Science Foundation of China (grant no. 31201586); Science and Technology Program of Guangdong, China (grant no. 2014A020208138 and 2015A020208018); Science and Technology Project of Zhanjiang City, China (grant no.2016A03016); Natural Science Foundation of Lingnan Normal University (grant no. LZL1507). All these funding play roles in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
Author information
Authors and Affiliations
Contributions
KL, HL, WL and JZ carried out the molecular studies, participated in the analysis and drafted the manuscript. YC carried out the qRT-PCR analysis. HL performed the statistical analysis. KL and CY conceived of the study, and participated in its design. KL acquired of funding. CS helped to draft the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The adult trees of A. squamosa cv. ‘Bendi’ are widely cultivated in China. This project uses plant materials and does not utilize transgenic technology. It does not require ethical approval.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
The detailed information of the obtained reads. (XLSX 8 kb)
Additional file 2:
GO classifications of unigenes. (XLSX 10 kb)
Additional file 3:
KEGG classifications of unigenes. (XLSX 13 kb)
Additional file 4:
The information of 701 TF genes were identified as DEGs. (XLSX 21 kb)
Additional file 5:
The information of 21 MADS-box genes were revealed from sugar apple flowers. (XLSX 15 kb)
Additional file 6:
Identification of key flowering- and flower development-related DEGs. (XLSX 12 kb)
Additional file 7:
The gene list of GO:0009908. (XLSX 10 kb)
Additional file 8:
Expression profiles of 15 key flower hormone-related genes during the flower development process. (DOCX 658 kb)
Additional file 9:
Expression profiles of eight floral organ development-associated TF-encoding genes during the flower development process. (DOCX 560 kb)
Additional file 10:
The primer sequences. (XLSX 10 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liu, K., Li, H., Li, W. et al. Comparative transcriptomic analyses of normal and malformed flowers in sugar apple (Annona squamosa L.) to identify the differential expressed genes between normal and malformed flowers. BMC Plant Biol 17, 170 (2017). https://doi.org/10.1186/s12870-017-1135-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-017-1135-y