
Abstract
Background
Enterocytozoon bieneusi is one of the commonest microsporidians contributing to human microsporidiosis, and is frequently found in animals in various countries. However, there is limited epidemiological information on this microorganism in Australia. Here, we undertook the first molecular epidemiological study of E. bieneusi in cats and dogs in Victoria.
Results
Genomic DNAs were extracted from 514 individual faecal deposits from cats (n = 172) and dogs (n = 342) and then tested using PCR-based sequencing of the internal transcribed spacer (ITS) of nuclear ribosomal DNA. Four distinct genotypes (designated D, PtEb IX, VIC_cat1 and VIC_dog1) of E. bieneusi were identified in 20 of the 514 faecal samples (3.9%). Genotype D is known to have a broad host range (humans and other animals) and has a wide geographical distribution around the world. The identification of this genotype here suggests that companion animals might represent reservoir hosts that are able to transmit E. bieneusi infection to humans in Australia. A phylogenetic analysis of ITS sequence data revealed that the novel genotype VIC_cat1 is related to the known genotype type IV within Group 1, and the new genotype VIC_dog1 is linked to a contentious “Group 3”, which includes genotypes reported previously in the published literature to represent Group 2 or 3.
Conclusions
A future, large-scale phylogenetic study of all known E. bieneusi genotypes, including VIC_dog1, should aid in clarifying their relationships and assignment to Groups, and in the identification of new genotypes, thus assisting epidemiological investigations.
Similar content being viewed by others

Background
Enterocytozoon bieneusi (Microsporidia) was first detected in 1985 in a Haitian patient (HIV/AIDS) suffering from severe diarrhoea [1]. As an emerging infectious agent, E. bieneusi is the commonest species contributing to human microsporidiosis [2], characterised by symptoms of acute or chronic diarrhoea, malabsorption and/or wasting [3]. Spores of E. bieneusi infect epithelial cells, replicate (proliferative phase) and are then shed as new, mature spores (sporogonic phase) in faeces into the environment; these spores can contaminate water (e.g., drinking source water and wastewater) and food, posing a risk to public health [3, 4]. Therefore, E. bieneusi has been classified as a Category B Priority Pathogen by the United States Environmental Protection Agency (EPA) [5].
It is not possible to specifically identify microsporidians using microscopic methods [6], such that PCR-based sequencing of the internal transcribed spacer (ITS) of nuclear ribosomal DNA is employed to identify E. bieneusi to the species and genotypic levels [4]. Using molecular tools, E. bieneusi has been found in a broad host range, including humans and various orders of other animals (including mammals: Artiodactyla, Lagomorpha, Carnivora, Perissodactyla, Primates and Rodentia, and birds: Columbiformes, Falconiformes, Galliformes and Passeriformes) as well as in water and food samples [3]. Some genotypes of E. bieneusi (e.g., D and EbpC) commonly reported in humans [7] have also been found in animals [8, 9]. This aspect raises the questions as to whether animals with a close affiliation with people play a significant role in the transmission of E. bieneusi infection to humans.
Studies of E. bieneusi in humans and other animals have been conducted in ~ 40 countries [10], but until recently, there has been very little research on this microsporidian in Australia. In 2018, we investigated E. bieneusi in wild herbivores. First, we identified some potentially zoonotic genotypes of E. bieneusi in wild sambar deer (D, J, MWC_d1-d2 and Type IV) [10] and marsupials (MWC_m1 and NCF2) inhabiting Melbourne’s water catchments (MWCs) [11]. Then, we extended our studies to farmed animals, including cattle in one of the MWCs (i.e. Tarago) [12] as well as alpacas in six states of Australia [13], to explore the prevalence and zoonotic potential of E. bieneusi in these farmed animals. The results from these studies revealed five genotypes (BEB4, I, J and TAR_fc1 and TAR_fc2) from cattle and one (ALP1) from alpaca with potential to infect humans. More recently, a study of E. bieneusi in humans [14] showed that genotype ALP1, previously found exclusively in alpaca [13], was identified in humans in Australia, indicating that this camelid may act as a reservoir for transmission to people.
In the present study, we investigated the prevalence of E. bieneusi in companion animals (cats and dogs) with a relatively close association to humans in both urban and rural environments in the state of Victoria, Australia, and characterised genotypes and assessed their zoonotic potential using a molecular phylogenetic approach.
Results
Using nested PCR-based sequencing of ITS, E. bieneusi DNA was detected in 20 of 514 (3.9%) faecal samples. Of these 20 test-positive samples, five (2.9% of 172) were from cats and 15 (4.4% of 342) from dogs (P = 0.478). Ten of them (7.9%; 10/126) were from juvenile cats (n = 4) and dogs (n = 6); and ten were from adults (2.7%; 10/371; including 1 cat and 9 dogs) (P = 0.016) (Tables 1 and 2). Of the 20 test-positive samples, nine (3.9%; 9/233) were from female cats (n = 2) and dogs (n = 7); and ten (3.7%; 10/270) were from male cats (n = 2) and dogs (n = 8), and one cat sample whose sex was not recorded (P = 1.000). Seven of these test-positive samples were collected in Spring and 13 in Summer (P = 0.494) (Table 2).
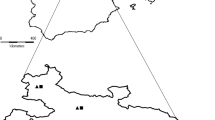
Most test-positive samples were from North Melbourne (n = 6), followed by Gladstone Park (~ 14 km from Lort Smith Animal Hospital), Toorak (8 km), and Woolamai (n = 2 each) (83 km), Collingwood (3 km), Glenroy (11 km), Northcote (5 km), Sunbury (36 km), Templestowe (17 km), Wildwood (27 km), Wyndham Vale (30 km), Yarraville (n = 1 each) (6 km), of which only Gladstone Park (n = 2), Sunbury (n = 1), Wildwood (n = 1), Woolamai (n = 2) and Wyndham Vale (n = 1) are not in the city of Melbourne (Fig. 1). There was no significant association of E. bieneusi prevalence with host species (P = 0.478), sex (P = 1.000) and season (P = 0.494). However, there was a significant association between age and prevalence for E. bieneusi (P = 0.016), with juvenile animals having 3.11-times higher risk of E. bieneusi-positivity than adults (OR = 3.11; 95% CI [1.264–7.663]) (Table 2).

Map indicating the geographical origins of individual cats and dogs from which faecal samples were collected in Victoria, Australia. Closed circles = locations from where faecal samples were collected. Blue open circles = Enterocytozoon bieneusi test-negative samples. Red open circles = E. bieneusi test-positive samples. Black solid circles = major cities in Victoria
The sequencing of the 20 individual ITS amplicons (241–244 bp) and their subsequent comparisons with reference sequences from the GenBank database revealed two known genotypes of E. bieneusi (D and PtEb IX) and two novel genotypes (called VIC_cat1 and VIC_dog1) (Table 3). The sequences determined unequivocally represented genotypes PtEb IX (50%; 10/20), D (40%; 8/20), followed by VIC_cat1 (5%; 1/20) and VIC_dog1 (5%; 1/20).
The ITS sequences from amplicons representing genotypes D (synonyms: CEbC, MJ10–12, NCF7, Peru9, PigEBITS9, PtEb VI, SHW1 and WL8) and genotype PtEb IX (synonyms: eb52 and EntcanA) were identical to those with accession nos. AF101200 (derived from human) (ITS; 243 bp) and DQ885585 (dog) (ITS; 244 bp), respectively (Additional file 2: Table S2). The ITS sequences from two amplicons representing VIC_cat1 (accession no. MK696086) and VIC_dog1 (accession no. MK696085) were one to two nucleotides different from the sequences with accession nos. AF242478 (type IV; human) and HM992519 (genotype CHN10; pig), respectively.
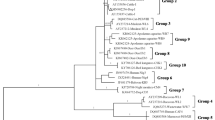
The ITS sequences of all four genotypes defined herein were aligned with sequences representing all ten established Groups of E. bieneusi [9, 15, 16], and then subjected to phylogenetic analysis (Fig. 2). In this analysis, Groups 1 to 10 were each strongly supported (pp = 0.95 to 1.00). Based on this analysis, genotype D and VIC_cat1 were assigned to Group 1 (pp = 1.00); genotype VIC_dog1 clustered with Group 3 (pp = 0.99); and genotype PtEb IX fell into Group 11 (outgroup) [14] with strong statistical support (pp = 1.00). Traditionally, E. bieneusi genotypes from dog, namely PtEb IX (accession no. DQ885585), CD7 (KJ668734) and CD8 (KJ668735), were commonly used as outgroup taxa in phylogenetic analyses, as their ITS sequences are quite distinct from E. bieneusi representing Groups 1–10.

Phylogenetic analysis of internal transcribed spacer (ITS) of nuclear ribosomal DNA sequence data (cf. Additional file 1: Table S1) by Bayesian inference (BI). Included here are ITS sequences of (i) E. bieneusi genotypes representing all currently recognised Groups (1 to 10) from the published literature, (ii) four genotypes of Enterocytozoon identified in the present study (bold-type) and (iii) the outgroup taxa. Statistically significant posterior probabilities (pp) of > 0.95 are indicated on branches. The scale-bar represents the number of substitutions per site. * = novel genotype
Discussion
This is the first molecular investigation of E. bieneusi from companion animals in Australia. Here, we established the genetic identity of E. bieneusi in faecal DNA samples from domestic cats and dogs using nested PCR-based sequencing of the ITS region. Globally, the prevalence of E. bieneusi in cats ranges from 1.4% (2/143) (household and stray) [17] to 31.3% (25/80) [18], while that in household and stray dogs ranges from 2.5% (2/79) [19] to 15.5% (54/348) [20] (Additional file 3: Table S3). By contrast, the prevalence of E. bieneusi in cats (2.9%) and dogs (4.4%) in our study was relatively low. The present study was designed to eliminate the possibility of genetically characterising E. bieneusi relating to endemic infections within an animal hospital environment - all faecal samples were collected from newly admitted animals. Therefore, the low prevalence of E. bieneusi in this study is plausible, as in other studies faecal samples were collected from pet markets or farms, where cats or dogs were raised together [16, 21], or from stray cats or dogs living under poor conditions [22]. Also important to consider is that various environmental factors (e.g., temperature and UV radiation), immunity, host age and sex, sample size and location may influence the prevalence rate.
Association analysis revealed that juvenile cats and dogs were significantly associated with a higher percentage of E. bieneusi prevalence than adults. This finding is supported by a previous study by Phrompraphai et al. [23], who conducted association analyses between E. bieneusi prevalence in dogs and their age, with dogs of less than one year of age having a higher prevalence of E. bieneusi infection. Some studies of E. bieneusi in other animals (e.g., cattle, deer, sheep and pig) [9, 24,25,26,27] and humans [14] have also revealed that age is a risk factor that might influence the prevalence of E. bieneusi infection. Host-age inversely associates with E. bieneusi prevalence, which could relate to immature immune systems in young animals. An association between E. bieneusi prevalence and geographical region could not be established due to an inadequate number of test-positive samples in individual localities.
The analysis of the ITS sequences derived from 20 faecal samples revealed four genotypes, including two known genotypes - D and PtEb IX (from both cats and dogs) - and two novel genotypes - VIC_cat1 and VIC_dog1. This is the first time that genotype PtEb IX, one of the most divergent genotypes of E. bieneusi, has been identified in Australia. An appraisal of current literature shows that PtEb IX appears to have a relatively narrow host-range, including dogs, cats and the European badger [28], with this genotype being principally found in dogs (Additional file 2: Table S2). This information suggests that genotype PtEb IX is transmitted predominantly among dogs, and is not likely to play a role in E. bieneusi transmission to humans. By contrast, genotype D is one of the commonest genotypes, and has a very broad host-range and a world-wide distribution [4]. This genotype has been found in more than 50 species of animals (Additional file 2: Table S2), including humans, as well as in water and foods, indicating that genotype D can be transmitted between humans and other animals (and vice versa), probably involving direct contact and/or the ingestion of E. bieneusi-contaminated water or food.
The identification of genotype D in cats and dogs in this study demonstrates that these companion animals likely serve as host reservoirs for selected E. bieneusi genotypes which are potentially transmissible to humans, particularly pet owners, due to close contact with their animals. Apart from genotype D, genotypes ALP3, Hum_q1–q3 and Ind4 (in the States of Queensland and Western Australia) [14] and genotype B (in the State of New South Wales) have been found in Australian people [2]. However, no studies of humans have yet been conducted in Victoria. Therefore, future studies of E. bieneusi in humans, especially those with and without pet cats and dogs, are warranted to establish whether “zoonotic” genotypes of E. bieneusi found in humans match those of their cats or dogs.
Phylogenetic analysis of the present ITS sequence data and selected sequences representing the ten established Groups of E. bieneusi (Fig. 2) revealed that novel genotype VIC_cat1 was most closely related to type IV, which has been frequently detected in humans and animals and represents Group 1, suggesting that genotype VIC_cat1 has zoonotic potential. However, the other novel genotype, VIC_dog1, that clustered with genotype CHN10, falls within a contentious “Group 3”, which includes genotypes reported previously to represent Group 2 or 3 [15, 16]. For instance, genotypes BEB4 and CHN10 have been recorded in Group 2, and genotypes S7 and WL4 have been placed in Group 3 (Additional file 1: Table S1). The ambiguous position of this novel genotype might lead to imprecise interpretations or conclusions regarding its epidemiological significance. Therefore, in our opinion, it would be useful to conduct a large-scale study of all unique ITS sequences (~ 600) reported and published to date, to search for patterns of nucleotide alterations in an alignment of all of these sequences, and to carry out a comprehensive phylogenetic analysis to untangle contradictions or confusions in the relationships of genotypes and Groups. Such an analysis would provide a refined framework for the assignment of new genotypes to Groups and would assist in assessing zoonotic potential in relation to members within Groups 2 to 10.
Conclusions
Exploring the genetic composition of E. bieneusi populations in animals and humans is important for understanding transmission patterns of disease (microsporidiosis), and for the prevention and control of this disease. By conducting the present molecular-phylogenetic investigation of E. bieneusi ITS rDNA sequences derived from faecal samples (n = 514) from household cats and dogs in Australia, we identified two known genotypes (D and PtEb IX) and two new genotypes VIC_cat1 and VIC_dog1. Genotypes D and VIC_cat1 both have zoonotic potential, suggesting that companion animals carrying these genotypes could be reservoirs for infection to humans. The novel genotype VIC_dog1 falls within a contentious Group, prompting the need for a future, large-scale molecular-phylogenetic analysis of all currently known E. bieneusi genotypes.
Methods
A total of 514 faecal samples from household cats (Felis catus; n = 172) and dogs (Canis lupus familiaris; n = 342), undergoing medical treatment, were donated by the Lort Smith Animal Hospital in North Melbourne between September 2018 and February 2019 (Table 1). In this hospital, fresh faecal samples were taken from newly admitted animals. Most of the animals (94.2%) were from the urban and suburban areas in Melbourne and environs, while others (5.1%) were from rural or regional towns in the State of Victoria (Fig. 1). Samples were from adult cats (n = 111) and dogs (n = 160); juvenile cats (n = 55) and dogs (n = 71); and from animals of unknown age (n = 17). Samples represented female cats (n = 72) and dogs (n = 161); male cats (n = 91) and dogs (n = 179); and a small number (n = 11) came from animals whose sex was not recorded (Table 1).
Genomic DNAs were extracted directly from 0.25 g of individual faecal samples using the PowerSoil kit (MoBio, USA), according to the manufacturer’s instructions. The ITS region of E. bieneusi was amplified from individual genomic DNAs by nested PCR using the primers MSP-1 (forward: 5′-TGA ATG KGT CCC TGT-3′) and MSP-2B (reverse: 5′-GTT CAT TCG CAC TAC T-3′) in the first round, and using primers MSP-3 (forward: 5′-GGA ATT CAC ACC GCC CGT CRY TAT-3′) and MSP-4B (reverse: 5′-CCA AGC TTA TGC TTA AGT CCA GGG AG-3′) in the second round [10]. Essential positive and negative controls were included in each PCR run.
Following column-purification, PCR products were directly sequenced [10]. The ITS sequences obtained in this study (publicly available under GenBank accession nos. MK696083–MK696086) were inspected for quality and compared with reference sequences acquired from the GenBank database (Additional file 1: Table S1). Genotypes of E. bieneusi were named in accordance with recommendations made by Santín and Fayer [4, 29].
All ITS sequences obtained from the present study, together with reference sequences were aligned, and subjected to phylogenetic analysis using using the Bayesian inference (BI) and Monte Carlo Markov Chain (MCMC) methods in MrBayes v.3.2.3 [10]. Posterior probability (pp) values were calculated by running 2,000,000 generations with four simultaneous tree-building chains, with trees being saved every one hundredth generation. A 50% majority rule consensus tree for each analysis was constructed based on the final 75% of trees generated by BI [10]. Genotypes were classified into groups using a recognised classification system [15, 16]. Chi-square and Fisher’s exact tests were utilised to test the association between E. bieneusi prevalence and possible risk factors (age, host, season and sex). The odds ratio (OR), calculated with a 95% confidence interval (95% CI), was used to measure the strength of association between the prevalence of E. bieneusi and a univariate risk factor. A P-value of < 0.05 was considered statistically significant. The SPSS Statistics package 25.0 (IBM, SPSS Inc., Chicago, IL) was used for all statistical analyses [14].
Availability of data and materials
Nucleotide sequences reported in this paper are available in the GenBank database under accession numbers MK696083–MK696086.
Abbreviations
- CI:
-
Confidence interval
- E. bieneusi :
-
Enterocytozoon bieneusi
- ITS :
-
Internal transcribed spacer of nuclear ribosomal DNA
- MWC:
-
Melbourne water catchments
- pp:
-
Posterior probability
References
Desportes I, Le Charpentier Y, Galian A, Bernard F, Cochand-Priollet B, Lavergne A, Ravisse P, Modigliani R. Occurrence of a new microsporidan: Enterocytozoon bieneusi n.g., n. sp., in the enterocytes of a human patient with AIDS. J Protozool. 1985;32(2):250–4.
Stark D, van Hal S, Barratt J, Ellis J, Marriott D, Harkness J. Limited genetic diversity among genotypes of Enterocytozoon bieneusi strains isolated from HIV-infected patients from Sydney, Australia. J Med Microbiol. 2009;58(3):355–7.
Santín-Durán M. Biology of foodborne parasites. In: Xiao L, Ryan UM, Feng Y, eidtors. Enterocytozoon bieneusi. 1st ed. Boca Raton, FL, USA: CRC Press; 2015. p. 149–174.
Santín M, Fayer R. Microsporidiosis: Enterocytozoon bieneusi in domesticated and wild animals. Res Vet Sci. 2011;90(3):363–71.
Didier ES, Weiss LM. Microsporidiosis: current status. Curr Opin Infect Dis. 2006;19(5):485–92.
Garcia LS. Laboratory identification of the microsporidia. J Clin Microbiol. 2002;40(6):1892–901.
Widmer G, Dilo J, Tumwine JK, Tzipori S, Akiyoshi DE. Frequent occurrence of mixed Enterocytozoon bieneusi infections in humans. Appl Environ Microbiol. 2013;79(17):5357–62.
Mynářová A, Foitová I, Kváč M, Květoňová D, Rost M, Morrogh-Bernard H, Nurcahyo W, Nguyen C, Supriyadi S, Sak B. Prevalence of Cryptosporidium spp., Enterocytozoon bieneusi, Encephalitozoon spp. and Giardia intestinalis in wild, semi-wild and captive orangutans (Pongo abelii and Pongo pygmaeus) on Sumatra and Borneo, Indonesia. PLoS One. 2016;11(3):e0152771.
Deng L, Li W, Zhong Z, Chai Y, Yang L, Zheng H, Wang W, Fu H, He M, Huang X. Molecular characterization and new genotypes of Enterocytozoon bieneusi in pet chipmunks (Eutamias asiaticus) in Sichuan province, China. BMC Microbiol. 2018;18(1):37.
Zhang Y, Koehler AV, Wang T, Haydon SR, Gasser RB. First detection and genetic characterisation of Enterocytozoon bieneusi in wild deer in Melbourne’s water catchments in Australia. Parasit Vectors. 2018;11(1):2.
Zhang Y, Koehler AV, Wang T, Haydon SR, Gasser RB. New operational taxonomic units of Enterocytozoon in three marsupial species. Parasit Vectors. 2018;11(1):371.
Zhang Y, Koehler AV, Wang T, Haydon SR, Gasser RB. Enterocytozoon bieneusi genotypes in cattle on farms located within a water catchment area. J Eukaryot Microbiol. 2018; https://doi.org/10.1111/jeu.12696. Accessed 25 Oct 2018.
Koehler AV, Rashid MH, Zhang Y, Vaughan JL, Gasser RB, Jabbar A. First cross-sectional, molecular epidemiological survey of Cryptosporidium, Giardia and Enterocytozoon in alpaca (Vicugna pacos) in Australia. Parasit Vectors. 2018;11:498.
Zhang Y, Koehler AV, Wang T, Robertson GJ, Bradbury RS, Gasser RB. Enterocytozoon bieneusi genotypes in people with gastrointestinal disorders in Queensland and Western Australia. Infect Genet Evol. 2018;65:293–9.
Karim MR, Dong H, Li T, Yu F, Li D, Zhang L, Li J, Wang R, Li S, Li X, et al. Predomination and new genotypes of Enterocytozoon bieneusi in captive nonhuman primates in zoos in China: high genetic diversity and zoonotic significance. PLoS One. 2015;10(2):e0117991.
Li W, Li Y, Song M, Lu Y, Yang J, Tao W, Jiang Y, Wan Q, Zhang S, Xiao L. Prevalence and genetic characteristics of Cryptosporidium, Enterocytozoon bieneusi and Giardia duodenalis in cats and dogs in Heilongjiang province, China. Vet Parasitol. 2015;208(3):125–34.
Li WC, Qin J, Wang K, Gu YF. Genotypes of Enterocytozoon bieneusi in dogs and cats in eastern China. Iran J Parasit. 2018;13(3):457–65.
Mori H, Mahittikorn A, Thammasonthijarern N, Chaisiri K, Rojekittikhun W, Sukthana Y. Presence of zoonotic Enterocytozoon bieneusi in cats in a temple in Central Thailand. Vet Parasitol. 2013;197(3):696–701.
Lobo ML, Xiao L, Cama V, Stevens T, Antunes F, Matos O. Genotypes of Enterocytozoon bieneusi in mammals in Portugal. J Eukaryot Microbiol. 2006;53(Suppl 1):61–4.
Karim MR, Dong H, Yu F, Jian F, Zhang L, Wang R, Zhang S, Rume FI, Ning C, Xiao L. Genetic diversity in Enterocytozoon bieneusi isolates from dogs and cats in China: host specificity and public health implications. J Clin Microbiol. 2014;52(9):3297–302.
Mathis A, Breitenmoser AC, Deplazes P. Detection of new Enterocytozoon genotypes in faecal samples of farm dogs and a cat. Parasite. 1999;6(2):189–93.
Santín M, Cortés Vecino JA, Fayer R. Enterocytozoon bieneusi genotypes in dogs in Bogota, Colombia. Am J Trop Med Hyg. 2008;79(2):215–7.
Phrompraphai T, Itoh N, Iijima Y, Ito Y, Kimura Y. Molecular detection and genotyping of Enterocytozoon bieneusi in family pet dogs obtained from different routes in Japan. Parasitol Int. 2019;70:86–8.
da Silva Fiuza VR, Lopes CW, de Oliveira FC, Fayer R, Santín M. New findings of Enterocytozoon bieneusi in beef and dairy cattle in Brazil. Vet Parasitol. 2016;216:46–51.
Li W, Diao R, Yang J, Xiao L, Lu Y, Li Y, Song M. High diversity of human-pathogenic Enterocytozoon bieneusi genotypes in swine in Northeast China. Parasitol Res. 2014;113(3):1147–53.
Santín M, Cortés Vecino JA, Fayer R. A zoonotic genotype of Enterocytozoon bieneusi in horses. J Parasitol. 2010;96(1):157–61.
Zou Y, Hou JL, Li FC, Zou FC, Lin RQ, Ma JG, Zhang XX, Zhu XQ. Prevalence and genotypes of Enterocytozoon bieneusi in pigs in southern China. Infect Genet Evol. 2018;44:52–6.
Santín M, Calero-Bernal R, Carmena D, Mateo M, Balseiro A, Barral M, Lima Barbero JF, Habela MÁ. Molecular characterization of Enterocytozoon bieneusi in wild carnivores in Spain. J Eukaryot Microbiol. 2018;65(4):468–74.
Santín M, Fayer R. Enterocytozoon bieneusi genotype nomenclature based on the internal transcribed spacer sequence: a consensus. J Eukaryot Microbiol. 2009;56(1):34–8.
Acknowledgements
The authors thank colleagues from Lort Smith Animal Hospital in North Melbourne for donating faecal samples.
Funding
Research funding from the Australian Research Council (ARC grant number LP160101299) and Melbourne Water Corporation (RBG and AVK) is gratefully acknowledged. The funding body (ARC) played no role in the design of the study, collection, analysis, or interpretation of data, or in the writing of the manuscript. Yan Zhang was the recipient of scholarships from the Chinese Scholarship Council (CSC) and The University of Melbourne.
Author information
Authors and Affiliations
Contributions
Design of Project: YZ, AVK, TW, DC and RBG. Acquisition of data: DC. Testing, analyses and interpretation: YZ, AVK, TW, DC and RBG. Wrote the paper: YZ, AVK and RBG. Supervision of project: AVK, TW and RBG. Grant funding: RBG and AVK. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. GenBank accession numbers of all internal transcribed spacer (ITS) of nuclear ribosomal DNA sequences used for phylogenetic analysis (Fig. 2), and associated information. Included here are ITS sequences of (i) E. bieneusi genotypes representing currently recognised Groups (1 to 10) from the published literature and genotypes without group assignment; (ii) four genotypes of Enterocytozoon identified/defined in the present study; and (iii) seven genotypes from the outgroups. (DOCX 94 kb)
Additional file 2:
Table S2. Genotypes D (synonyms: CEbC, MJ10-12, NCF7, Peru9, PigEBITS9, PtEb VI, SHW1and WL8) and genotype PtEb IX (synonyms: eb52 and EntcanA) of Enterocytozoon bieneusi recorded in different host species, food and water samples fromprevious publications. (DOCX 284 kb)
Additional file 3:
Table S3. All Enterocytozoon bieneusi genotypes, prevalence and risk factors recorded previously in cats (Felis catus) and dogs (Canis lupus familiaris) worldwide. (DOCX 75 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, Y., Koehler, A.V., Wang, T. et al. Enterocytozoon bieneusi genotypes in cats and dogs in Victoria, Australia. BMC Microbiol 19, 183 (2019). https://doi.org/10.1186/s12866-019-1563-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-019-1563-y