
Abstract
Background
We performed a 12-month cohort study of the stability and resilience of the intestinal microbiota of healthy children in daycare in Denmark in relation to diarrheal events and exposure to known risk factors for gastrointestinal health such as travelling and antibiotic use. In addition, we analyzed how gut microbiota recover from such exposures.
Results
We monitored 32 children in daycare aged 1–6 years. Fecal samples were submitted every second month during a one-year observational period. Information regarding exposures and diarrheal episodes was obtained through questionnaires. Bacterial communities were identified using 16S rRNA gene sequencing. The core microbiota (mean abundance > 95%) dominated the intestinal microbiota, and none of the tested exposures (diarrheal events, travel, antibiotic use) were associated with decreases in the relative abundance of the core microbiota. Samples exhibited lower intra-individual variation than inter-individual variation. Half of all the variation between samples was explained by which child a sample originated from. Age explained 7.6–9.6% of the variation, while traveling, diarrheal events, and antibiotic use explained minor parts of the beta diversity. We found an age-dependent increase of alpha diversity in children aged 1–3 years, and while diarrheal events caused a decrease in alpha diversity, a recovery time of 40–45 days was observed.
Among children having had a diarrheal event, we observed a 10x higher relative abundance of Prevotella. After travelling, a higher abundance of two Bacteroides species and 40% less Lachnospiraceae were seen. Antibiotic use did not correlate with changes in the abundance of any bacteria.
Conclusion
We present data showing that Danish children in daycare have stable intestinal microbiota, resilient to the exposures investigated. An early age-dependent increase in the diversity was demonstrated. Diarrheal episodes decreased alpha diversity with an estimated recovery time of 40–45 days.
Similar content being viewed by others

Background
The healthy human intestinal bacterial microbiota has been described as a diverse microbial community dominated by Firmicutes, Bacteroidetes and Actinobacteria and to a lesser extent Proteobacteria and Verrucomicrobia [1]. Various exposures, including foreign travel, consumption of antibiotics and environmental factors have been shown to greatly influence the structure of the intestinal microbiota [2,3,4,5,6]. Likewise, the composition and diversity of the human intestinal microbiota has been associated with various gastrointestinal diseases [7,8,9,10,11]. Taxa with a relatively high abundance such as Ruminococcaceae, Enterobacteriaceae, Fusobacteriaceae, Pasteurellaceae and Veillonelleceae have been associated with Crohn’s disease [7, 12] and Desulfovibrio with ulcerative colitis [13]. Meanwhile a ‘healthy microbiota’ has been associated with an increased abundance of Faecalibacterium prautsnitzii, Escherichia coli, Oscillibacter spp. and Bifidobacterium [14,15,16,17]. Furthermore, decreased bacterial alpha diversity has been associated with several gastrointestinal diseases [12, 18,19,20].
The emergence of new technologies has allowed for a deeper understanding of the interplay between bacterial communities in the gut, e.g., when challenged by diarrheal episodes and in the following period of recovery [14, 21, 22]. High-throughput 16S rRNA gene sequencing has provided an opportunity for a more holistic approach to monitoring microbial interactions in health and disease compared with conventional culturing of fecal samples, where the focus has been on identifying single enteric pathogens as causes of disease. By shifting the focus from individual bacterial species to studying entire microbial communities, it is possible to investigate and gain insight into how complex the alterations in the microbiota are in periods of health and disease.
Here, we present the first longitudinal study investigating compositional changes in the intestinal microbiota of children in daycare before and after diarrheal events and exposure to known risk factors (e.g., travel and use of antibiotics). Thus, we come a step closer to understand the dynamics of the intestinal microbiota in otherwise healthy children. In addition, we aim to define specific bacterial taxa that might confer an increased colonization resistance or greater resilience towards development of diarrhea.
Results
We investigated the intestinal microbiome of 32 healthy children, with a median age of two years, for a period of one year. Our longitudinal observations enabled analysis of paired and non-paired data pertaining to the dynamics in the intestinal microbiota in Danish children in daycare.
Sequencing depth
We created rarefaction curves for both observed richness and Shannon Diversity Index (H′) in order to determine the minimum sequencing depth that properly described the microbiota (Additional file 1: Figure S1). The observed richness was dependent on sequencing depth, but the rate of increase became significantly lower when including more than 2000 reads. The H′ reached a plateau when including more than 1000 reads. Consequently, we excluded 10 samples with less than 2000 reads for further analysis, and rarefied the remaining samples to 3500 reads per sample. This rarefied dataset was used for all analyses in the study.
Stability of the microbiota
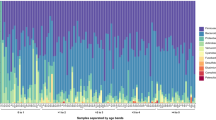
To identify the stability of the microbiota, we identified the core microbiota for each individual child (defined as the operational taxonomic units (OTUs) present in all samples from each individual child). The median relative abundance of the core microbiota in each sample was 95.9% (interquartile range (IQR): 92.6–97.4%, Additional file 2: Figure S2) and did not correlate with diarrheal episodes, antibiotic treatment, travelling (in or outside Scandinavia), or any combination of these perturbations.
Beta diversity
Using both Bray-Curtis and unweighted UniFrac distance metrics, we observed that samples differed more between children than within children (Fig. 1, Wilcoxon, p < 10− 15).

Boxplot showing intra- (green) and inter- (red) individual variation in gut microbiota over time according to age. The box-plot ranges from the 25th to the 75th percentile, with the 50th percentile represented by the black horizontal line
As verification of our findings, we used permutational multivariate analysis of variance using distance matrices (PERMANOVA) to determine the amount of the overall variation in beta diversity explained by the individual child. Based on Bray-Curtis distances and unweighted UniFrac distances the individual child explained 53.6 and 48.3% of the overall variation, respectively (p < 0.001).
The effect of age, travelling, diarrheal episodes, and antibiotic use on the microbial composition was tested using PERMANOVA, for both distance metrics (full results in Additional file 3: Table S1). The largest part of the variation was explained by the age of the child, 7.6% in the Bray-Curtis distances (p < 0.001). When testing the variation in the Bray-Curtis distances separately, travelling significantly explained 2.1% (p = 0.002), diarrhea 1.4% (p = 0.015), while antibiotic use did not significantly explain any of the overall variation (1.3%, p = 0.22). When all four variables were tested sequentially in one model (ordered as above), varying parts of the overall variation was explained by age (7.6%, p < 0.001), travelling (1.8%, p = 0.002), and diarrhea (1.2%, p = 0.014), whereas antibiotic use did not contribute significantly to explaining the variation observed in the samples. Furthermore, episodes of diarrhea explained additional 6.0% of the variation within each age group (p = 0.001).
For the unweighted UniFrac distances, the age of the child at sampling correspondingly explained the largest part of the variation (9.3%, p = 0.001). On the other hand, antibiotic use explained 2.1% (p = 0.001), while both travelling and diarrhea explained 1.4% (p = 0.002 and p = 0.001, respectively). When including all variables in the analysis (in the same order as above), antibiotics explained 1.2% (p = 0.003), travelling 1.4% (p = 0.001), and diarrhea 1.1% (p = 0.012) of the overall variation. Regarding variation within groups, travelling explained additional 3.7% of the overall variation within the age groups (p = 0.009) and 0.9% within the antibiotic treatments (p = 0.047). No other combination of variables significantly explained any of the remaining variation.
Alpha diversity
To test if the four variables (age, travelling, diarrhea, and antibiotic use) had an impact on the diversity of the individual samples, we investigated any correlation between the variables and the alpha diversity using both observed richness and H′. The observed richness of each sample indicated no temporal trend within each child (Fig. 2a). When grouping the samples by child age, we observed a significant correlation between age at sampling and observed richness (analysis of variance (ANOVA), p < 10− 4, Tukey Honest Significant Differences (TukeyHSD): 1 year lower than 2–6 years, p = 0–0.003, Fig. 2b), with a similar trend for H′ (ANOVA, p < 10− 7, TukeyHSD: 1 year lower than 3–6 year, p = 0.0003–0.037, data not shown). Furthermore, for children with a diarrheal event within the last 60 days, the alpha diversity could be explained as a function of the time passed after the reported diarrheal episode. To investigate this, we used generalized linear models (glm) (observed richness: glm, p < 10− 10, Fig. 2c. H′: glm, p < 10− 13, Fig. 2d). Comparing these models to the mean observed richness (55.76) and H′ (2.59) in samples where the child had not had a diarrheal events in the two months prior, the observed richness recovered after 39.86 days and H′ recovered after 44.75 days.

Alpha diversity. a Observed richness of each sample grouped for each child and colored by sampling time. b Boxplot of the observed richness grouped by the age at sampling. c The observed richness and D) H′ as function of days since last diarrheal episode (dots), with the blue line showing a glm and the grey area the 95% confidence interval
Effects of diarrhea, travel and antibiotic use on single bacteria abundances
We used a permutational test to determine the significance of any differences in abundance of specific bacterial taxa following diarrheal episodes, travel, and antibiotic usage. We included all OTUs with a minimum average abundance of 0.05%, and performed the analysis at all taxonomical levels. The test was performed for diarrhea (23.8%, 41 of 172 samples [9 NA]), travelling (27.0%, 47 of 174, [7 NA]), and antibiotic use (7.0%, 12 of 172 [9 NA]). Across all taxonomic ranks we identified 51 (Antibiotic use: 14, Travelling: 18, Diarrhea: 19) association with p < 0.05, but only 12 (Travelling: 9, Diarrhea: 3) survived correction for false discovery rate (Fig. 3, Additional file 3: Table S2).

Vulcano plot of the differential abundant bacteria. X-axis reflects the log10 value of fold change in abundance for each variable (exposed no/yes). Y-axis is -log10 of the adjusted p-values. The shape indicates the exposure (Antibiotics: Circle, Diarrhea: Triangle, Travelling: Square) and the color indicates the taxonomic rank from phylum to species level
Children who had experienced at least one diarrheal episode within the last two months, had an approximately 10-fold increase of the genus Prevotella (2.8% vs. 0.28%, adjusted p-value = 0.01).
Children, who had travelled within a period of two months prior to sampling had an increased relative abundance of two Bacteroides species, B. bacterium_NLAE-zl-P191 and B. thetaiotaomicron, (1.1% vs. 0.65 and 2.1% vs. 0.81%, respectively, adjusted p-values = 0.008). Furthermore, a decrease of the family Lachnospiraceae was observed (9.1% vs. 14.8%, adjusted p-value = 0.009).
Antibiotic usage did not cause any significant changes in relative abundances of the bacteria in our data set.
We tested if any genera, with a mean read count higher than 1 per sample, correlated with the alpha diversity. Using the DAtest function, we estimated the efficiency of all linear models (data not shown) and based on these data, we selected both linear regression modelling (false discovery rate: 0.056, true positive rate: 0.333) and log linear regression modelling (false discovery rate: 0.040, true positive rate: 0.200). Fourteen genera were determined to have a significant correlation with both H′ and observed richness, using both methods. The majority was observed to be positively correlated with alpha diversity (Fig. 4, Additional file 3: Table S3). The only negatively correlated genus was Bacteroides, which is the most abundant genus in this study (mean: 32.7%, IQR: 22.5–43.3%).

Correlation between alpha diversity and bacterial abundance. Alpha diversity plotted as a function of abundance in each sample, with observed richness (red) scale on the left y axis and H′ (blue) on the right y axis. The lines represent a general linear model with the grey area showing 95% confidence intervals
Discussion
We found that children in daycare had a stable intestinal microbiota, resilient to the perturbations investigated. The core microbiota in each child dominated their microbiota, above 95% mean relative abundance, throughout the study; none of the tested exposures correlated with a decrease in the relative abundance of the core microbiota in general, nor when looking at each child separately (Additional file 2: Figure S2). The analysis of beta diversity variation showed a greater similarity between samples collected from the individual child compared with samples collected from other children (Fig. 1), which further highlights the considerable individual contribution to the unique composition of the microbiota even in small children. In addition, the individual child from which a sample was collected explained approximately half of all variation between samples. This was observed using both the Bray-Curtis distance metric, a merely abundance-based approach, and unweighted UniFrac distances, which is a distance metric based on the phylogenetic similarity between the bacteria present in the samples.
Age at sampling was a significant factor for the variation observed between samples; it explained additional 7.6–9.6% of the variation, supporting earlier studies, and showing how the intestinal microbiota is still undergoing some maturation at this young age [23,24,25]. Travel interestingly explained more of the variation, when using a purely abundance-based distance metric (2.1% by Bray-Curtis), as opposed to the comparison of the phylogenetic similarities between the bacteria present in the samples (1.4%, unweighted UniFrac). The difference in explained variance could indicate that microbiota changes associated with travel relates to a shift in the relative abundance of the bacteria present, or introduction of closely related bacteria, and to a lesser extent to introduction of foreign bacteria. Diarrheal events only explained 1.4% of the variation, using either distance metric, which was less than expected, when considering the association between stool consistency and microbiota composition [26]. Antibiotic usage explained more of the variation, when using unweighted UniFrac (2.1%) compared with Bray-Curtis (1.3%); it is interesting to note that only the presence/absence based unweighted UniFrac distance explained a significant variation from antibiotic usage. This difference in the amount of variation beta-diversity explained for the two distance metrics suggest that antibiotic use affects low-abundant bacteria, which might have been removed from the child’s microbiota, and to a lesser effect on the dominant bacteria. To speculate any further on the low variation explained by travelling, diarrhea, or antibiotic use should be subject to caution, due to the varying time intervals between antibiotic use and sample collection in our study. The fixed sampling interval meant that the time between diarrheal events and the sample collection ranged from 0 to 59 days. This large variation in the time passed since diarrheal event could explain this discrepancy, as it is difficult to differentiate between the direct impact of diarrheal events and the microbiota resilience, the ability of the microbiota to reestablish itself.
Looking at the alpha diversity characteristics, observed richness and H′, no time-dependent patterns within each child were observed. However, an overall increase in alpha diversity was observed in the younger children, from 1 to 3 years of age (Fig. 1a, b). In addition to the age-dependent trend in alpha diversity, we found a clear indication of alpha diversity being lower after a diarrheal event and that a time-dependent increase in alpha diversity related to the number of days since the diarrheal event took place; this development was clearest when analyzing H′ (Fig. 1c, d). When comparing alpha diversity/time correlation to samples without reports of diarrheal episodes, we could estimate that the alpha diversity reached the mean level of children without diarrheal episodes after 40–45 days. This dose-dependent effect in means of days past since a diarrheal event indicates a strong correlation between diarrhea and alpha diversity in the microbiota of small children.
In an effort to identify any more specific impact of travelling, diarrheal events and antibiotic use on the microbiota, we performed a permutation test to identify bacteria, with a different relative abundance between groups (Fig. 4). Prevotella was significantly increased following diarrheal events; we saw a 10x higher average relative abundance in children having had a diarrheal event. Such an increase does not necessarily implicate Prevotella as a causative agent in the diarrheal episode, but might suggest early re-colonization of Prevotella in the period of recovery after a perturbation of the microbiota caused by diarrhea. This is likely to be the first step in the succession of re-colonization events, and has also been described as one of the stages after Vibrio cholera-induced childhood diarrhea, where increased abundance of Prevotella is associated with late-stage recovery from infectious diarrhea and interpreted as a return to a healthy gut microbiota [27].
In children, who had been travelling, we saw an increase of two Bacteroides species, B. thetaiotaomicron (2.6 times higher) and B. bacterium_NLAE-zl-P191 (1.7 times higher). In these children, we observed a 40% decrease in the family Lachnospiraceae, a member of the class Clostridia. In general, the phylum Bacteroidetes tended to be increased by 16% after travelling (not statistically significant), while the phylum Firmicutes was decreased by 16%. This change in bacterial abundance might be associated with changes in diet during travelling. No significantly differential abundant bacteria were seen in relation to antibiotic use.
Lastly, we looked at possible correlations between alpha diversity and abundance of individual bacterial genera. We found 13 genera positively correlated with alpha diversity, and only one (Bacteroidetes) was negatively correlated with alpha diversity (Fig. 4). That the most abundant genus in the entire dataset is negatively correlated with alpha diversity is not surprising, since a high relative abundance of a single OTU will decrease the number of other OTUs found within a given number of observations.
This longitudinal cohort study of healthy children aged 1–6 years with samples collected over a 12-month period enabled us to monitor the intestinal microbiota of children in daycare from a perspective never presented before. As a study based on the use of 16S rRNA gene amplicon sequencing the strengths and limitations of the technique is inherent in this study. The high sequencing depth and number of reads per sample give a very good foundation for the multiple statistical tests used, and, even more importantly, it allowed us to identify bacteria in different phases of the child’s life, through disease and health, which would be difficult to identify using a classical culture-dependent approach. Fecal culturing for microbial pathogens was performed and described earlier [16]. The findings reflected that the children were carriers of a numerous pathogens during the observation year. No correlations throughout the year for each child could be found by culturing.
The main limitation of this study is the level of resolution of the data, as we have amplified and sequenced the V4 region of the 16S rRNA gene followed by clustering of reads into OTUs with a 97% identity threshold. Therefore, many OTUs will contain multiple species, and even genera, as these may differ by less than 3% within the V4 region. Furthermore, as we cannot even differentiate species, this also means that differentiation between e.g., a commensal E. coli and a pathogenic enterotoxin-producing E. coli was not possible. Our study design, with samples collected at fixed 2 month intervals, provided a unique opportunity to compare samples before and after perturbation of the microbiota. Lastly, any partial recovery of the gut microbiota occurring after any exposure will have obscured the effect of the exposure due to the variable intervals of time between sampling.
Conclusion
We here present a study of the fecal microbiota of children in Danish daycare, observed over a 12-month period, and the impact of three types of exposures for each child: Travelling, diarrhea, and antibiotic use. The fecal microbiota of each child were dominated by a stable core microbiota, and the abundance of the core microbiota were not affected by the exposures investigated. While a few specific bacteria were affected by travelling and diarrhea. This indicate that already at this early age, contradictory to the common conception, the children have a very early establishment of a highly individualized and robust fecal microbiota community. We did observe an age-dependent increase in alpha diversity of the intestinal microbiota; this was seen strongest in the youngest children (1–3 years of age).
Most interestingly, our analysis of the alpha diversity, following diarrheal events, showed a clear decrease in alpha diversity and an estimated recovery period of 40–45 days.
Methods
Subjects and samples
A cohort of 179 healthy children aged 1 to 6 years was established to investigate the incidence of enteric pathogens in relation to diarrheal episodes in daycare in Denmark. The cohort were created to investigate incidence and clinical significance of Enteroaggregative Escherichia coli in healthy Danish children in daycare and the primary results, using conventional microbiological tests for enteric pathogens, have been published [16, 17, 28, 29].
The study was designed as a dynamic cohort study, where each child was included for a one-year period with a follow-up point every second month with submission of a fecal sample and a questionnaire. The questionnaire inquired about gastrointestinal symptoms, use of antibiotics and various exposures in relation to gastrointestinal disease, and was completed by the parents. In total, 2160 children were invited to participate in the cohort, 200 (9.6%) accepted the invitation, and of these 175 (87.5%) submitted at least one sample. Of these 32 (18.3%) submitted 5 (1) or 6 (31) fecal samples, and the 191 samples from these children were analyzed using 16S rRNA gene amplicon sequencing (Fig. 5).

Flow chart of participating children. A cohort of healthy children aged 1 to 6 years was established. Each child was included for a one-year period with a follow-up point every second month with submission of a fecal sample and a questionnaire. In total, 2160 children were invited to participate in the cohort, 200 (9.6%) accepted the invitation, and of these 175 (87.5%) submitted at least one sample. Of these 32 (18.3%) submitted 5 (1) or 6 (31) fecal samples, and the samples (n = 191) from these children were analyzed using 16S rRNA gene amplicon sequencing
DNA extraction
Fecal samples were collected by at home and shipped to Statens Serum Institut, Copenhagen. The fecal was transferred to Eppendorf® tubes and kept at − 80 °C. Bacterial DNA was extracted from fecal material using the Qiagen QIAmp DNA Stool Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. Briefly, 200 mg fecal material was mixed with 1.4 mL ASL buffer and vortexed until homogenization. In the next step, 0.2 g sterile zirconia/silica beads (diameter 0.01 mm, BioSpec Products ROTH Karlsruhe, Germany) were added, and samples were processed on the Tissuelyzer (Qiagen Retsch GmbH, Hannover, Germany) for 6 min at 30 Hz. This was followed by lysis for 5 min at 95 °C. The final sample preparation was eluted in 100 μL buffer [30,31,32].
PCR and Miseq analysis
16S rRNA gene amplification was performed using a nested two-step protocol, targeting the hypervariable V4 region of the 16S rRNA gene. In the first step, the V3 and V4 regions were amplified using the modified broad-range primers Uni340F (5’ ̶ CCT AYG GGR BGC ASC AG ̶ 3′) and Uni806R (5’ ̶ GGA CTA CNN GGG TAT CTA AT ̶ 3′) [33,34,35,36]. Amplification was performed in 96-well microtiter plates with a reaction mixture consisting of 1X AccuPrime PCR Buffer II, 0.6 U AccuPrime Taq DNA Polymerase (Invitrogen, Life technologies, CA, US), 0.5 μM primer 341F, 0.5 μM primer 806R, and 2.0 μL template DNA, resulting in a total volume of 20.0 μL for each sample. The first PCR step was performed using the following program: 2 min at 94 °C (denaturation), 30 cycles of 20 s at 94 °C (denaturation), 30 s at 56 °C (annealing) and 40 s at 68 °C (elongation), and lastly 5 min at 68 °C (final extension). A negative template-free control and a positive control were applied for each plate containing 2.0 μL DNA from a known bacterial mock community (1.0 ng/μL; HM-782D, BEI Resources, VA, US). PCR products were quantified using the Quant-iT PicoGreen quantification system (Life Technologies, CA, US). Prior to further analysis, dilution of samples with a DNA concentration of more than 6.0 ng/μL was performed to achieve a concentration of approximately 3.0–6.0 ng/μL.
In the second PCR step, we targeted the V4 region while also adding sequencing primers and adaptors to the amplicon products. This step was performed as above, with 2.0 μL of the diluted amplicon products and sequence-specific primers, with sequencing adaptors and unique index combinations for each sample as described by Mortensen et al. 2016 [37]. In addition, the number of PCR cycles was reduced to 15. Amplicons were purified with Agencourt AMPure XP Beads (Beckman Coulter Genomics, MA, US) according to the manufacturer’s instructions using 0.7X volume beads and quantified as previously described. Equimolar amounts of the amplification products were pooled in a single tube. Pooled DNA samples were concentrated using the DNA Clean & ConcentratorTM-5 Kit (Zymo Research, Irvine, CA, US) according to the manufacturer’s instructions, and the final DNA concentration of the pooled library was determined using the Quant-iT™ High-Sensitivity DNA Assay Kit (Life Technologies) according to the instructions of the manufacturer. Amplicon sequencing was performed on the Illumina MiSeq Desktop Sequencer (Illumina Inc., CA, US). The samples were sequenced using the MiSeq Reagent Kits v2 (Illumina Inc., CA, US), and 5.0% PhiX was added as internal control. We performed 250 paired-end sequencing with dual-index reads.
For the development of OTU tables, taxonomic assignment, and phylogenetic analysis, we used our in-house bioinformatics pipeline [38]. Briefly, the pipeline used several tools to perform trimming (bioDSL [https://github.com/maasha/BioDSL]), mate pairing and quality filtering (usearch v7.0.1090 [39]), OTU clustering and singleton removal (97%, UPARSE [40]), chimera checking against the GOLD Database [41], taxonomical classification of representative sequences (Mothur v.1.25.0 [42]), construction of phylogenetic tree (PyNAST [43], FastTree [44], and filter_alignment.py [45]), and lastly, alignments against the 2011 version of Greengenes [46].
Data analysis
Statistical analysis was performed in R (v3.4.1) [47], using several R packages. Import of the sequencing data (OTU table, taxonomic table, and phylogenetic tree) and sample information into R and general analysis were performed using the phyloseq package [48]. We used two measures of alpha diversity: observed richness (the number of unique OTUs found within each sample) and Shannon Diversity Index (H′) (a composite measure of the bacterial richness and the evenness of the relative bacterial abundance within each sample); both measures were calculated using phyloseq functionality. Using phyloseq functions we created rarefaction curves, determined a minimum sequencing depth, removed insufficiently sequenced samples and created a rarefied OTU table used for further analysis. For comparison of the overall compositional similarities and differences between the samples (beta diversity), we used two different distance metrics: Bray-Curtis similarity (based on relative abundances) and unweighted UniFrac distance (based on the phylogenetic differences between OTUs present or absent in the samples). In order to test how much of the overall variation in the microbial composition that could be explained by one or multiple categorical variables, PERMANOVA was performed using the function “adonis” from the R package Vegan [49]. Differences in alpha diversity between categorical variables were tested with ANOVA, followed by Tukey’s honest significant difference test (TukeyHSD) for between-group variance. Correlations with continuous variables were tested using generalized linear models (glm), all three using the R package stats. Differential abundance testing was performed using the test described by Thorsen et al. (2016) [50], the test perform between group comparison for all bacteria and includes false discovery rate (FDR) adjustment of p-values. Additional functions from the DAtest R package [51] were used to analyze correlations between the relative abundance of bacteria and alpha diversity. All plots were created using the R package ggplot2 [52].
Abbreviations
- ANOVA:
-
Analysis of variance
- glm:
-
Generalized linear models
- H′:
-
Shannon diversity index
- IQR:
-
interquartile range
- OTU:
-
Operational taxonomic unit
- PERMANOVA:
-
Permutational multivariate analysis of variance using distance matrices
- TukeyHSD:
-
Tukey honest significant differences
References
Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. 2014;146:1489–99.
Becker-Dreps S, Allali I, Monteagudo A, Vilchez S, Hudgens MG, Rogawski ET, et al. Gut microbiome composition in young Nicaraguan children during diarrhea episodes and recovery. Am J Trop Med Hyg. 2015;93:1187–93.
Deshmukh HS, Liu Y, Menkiti OR, Mei J, Dai N, O’Leary CE, et al. The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nat Med. 2014;20:524–30.
Flint HJ, Scott KP, Louis P, Duncan SH. The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol. 2012;9:577–89. https://doi.org/10.1038/nrgastro.2012.156.
Hsiao A, Ahmed AMS, Subramanian S, Griffin NW, Drewry LL, Petri WA, et al. Members of the human gut microbiota involved in recovery from Vibrio cholerae infection. Nature. 2014;515:423–6. https://doi.org/10.1038/nature13738.
Monira S, Nakamura S, Gotoh K, Izutsu K, Watanabe H, Alam NH, et al. Metagenomic profile of gut microbiota in children during cholera and recovery. Gut Pathog. 2013;5(1).
Joossens M, Huys G, Cnockaert M, De Preter V, Verbeke K, Rutgeerts P, et al. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut. 2011;60:631–7. https://doi.org/10.1136/gut.2010.223263.
Rowan F, Docherty NG, Murphy M, Murphy B, Coffey JC, O’Connell PR. Desulfovibrio bacterial species are increased in ulcerative colitis. Dis Colon Rectum. 2010;53:1530–6.
Mondot S, Kang S, Furet JP, Aguirre De Carcer D, McSweeney C, Morrison M, et al. Highlighting new phylogenetic specificities of Crohn’s disease microbiota. Inflamm Bowel Dis. 2011;17:185–92.
Miquel S, Martín R, Rossi O, Bermúdez-Humarán LG, Chatel JM, Sokol H, et al. Faecalibacterium prausnitzii and human intestinal health. Curr Opin Microbiol. 2013;16:255–61.
Tojo R, Suárez A, Clemente MG, De los Reyes-Gavilán CG, Margolles A, Gueimonde M, et al. intestinal microbiota in health and disease: role of bifidobacteria in gut homeostasis. World J Gastroenterol. 2014;20:15163–76.
Kang D-W, Park JG, Ilhan ZE, Wallstrom G, LaBaer J, Adams JB, et al. Reduced incidence of Prevotella and other fermenters in intestinal microflora of autistic children. PLoS One. 2013;8(7):e68322.
Vincent C, Stephens DA, Loo VG, Edens TJ, Behr MA, Dewar K, et al. Reductions in intestinal Clostridiales precede the development of nosocomial Clostridium difficile infection. Microbiome. 2013;1:18. https://doi.org/10.1186/2049-2618-1-18.
Samanta AK, Torok VA, Percy NJ, Abimosleh SM, Howarth GS. Microbial fingerprinting detects unique bacterial communities in the faecal microbiota of rats with experimentally-induced colitis. J Microbiol. 2012;50:218–25.
Scanlan PD, Shanahan F, Marchesi JR. Culture-independent analysis of desulfovibrios in the human distal colon of healthy, colorectal cancer and polypectomized individuals. FEMS Microbiol Ecol. 2009;69:213–21.
Hebbelstrup Jensen B, Stensvold CR, Struve C, Olsen KEP, Scheutz F, Boisen N, et al. Enteroaggregative Escherichia coli in daycare—a 1-year dynamic cohort study. Front Cell Infect Microbiol. 2016;6. https://doi.org/10.3389/fcimb.2016.00075.
Hebbelstrup Jensen B, Röser D, Andreassen BU t, Olsen KEP, Nielsen HV e, Roldgaard BB j, et al. Childhood diarrhoea in Danish day care centres could be associated with infant colic, low birthweight and antibiotics. Acta Paediatr. 2016;105:90–5. https://doi.org/10.1111/apa.13209.
Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148:1258–70.
Collado MC, Cernada M, Baüerl C, Vento M, Pérez-Martínez G. Microbial ecology and host-microbiota interactions during early life stages. Gut Microbes. 2012;3:352–65.
David LA, Materna AC, Friedman J, Campos-Baptista MI, Blackburn MC, Perrotta A, et al. Host lifestyle affects human microbiota on daily timescales. Genome Biol. 2015;15:R89.
Ng SC, Lam EFC, Lam TTY, Chan Y, Law W, Tse PCH, et al. Effect of probiotic bacteria on the intestinal microbiota in irritable bowel syndrome. J Gastroenterol Hepatol. 2013;28(10):1624–31. https://doi.org/10.1111/jgh.12306.
Guarner F, Malagelada J-R. Gut flora in health and disease. Lancet. 2003;361:512–9. https://doi.org/10.1016/S0140-6736(03)12489-0.
Bergström A, Skov TH, Bahl MI, Roager HM, Christensen LB, Ejlerskov KT, et al. Establishment of intestinal microbiota during early life: a longitudinal, explorative study of a large cohort of Danish infants. Appl Environ Microbiol. 2014;80:2889–900. https://doi.org/10.1128/AEM.00342-14.
Laursen MF, Zachariassen G, Bahl MI, Bergström A, Høst A, Michaelsen KF, et al. Having older siblings is associated with gut microbiota development during early childhood. BMC Microbiol. 2015;15:154. https://doi.org/10.1186/s12866-015-0477-6.
Stokholm J, Blaser MJ, Thorsen J, Rasmussen MA, Waage J, Vinding RK, et al. Maturation of the gut microbiome and risk of asthma in childhood. Nat Commun. 2018;9(1):704.
Vandeputte D, Falony G, Vieira-Silva S, Tito RY, Joossens M, Raes J. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut. 2016;65:57–62.
David LA, Weil A, Ryan ET, Calderwood SB, Harris JB, Chowdhury F, et al. Gut microbial succession follows acute secretory diarrhea in humans. MBio. 2015;6:e00381–15. https://doi.org/10.1128/mBio.00381-15.
Jokelainen P, Hebbelstrup Jensen B, Andreassen BU, Petersen AM, Röser D, Krogfelt KA, et al. Dientamoeba fragilis, a commensal in children in Danish day care centers. J Clin Microbiol. 2017;55:1707–13. https://doi.org/10.1128/JCM.00037-17.
Sydenham TV, Jensen BH, Petersen AM, Krogfelt KA, Justesen US. Antimicrobial resistance in the Bacteroides fragilis group in faecal microbiota from healthy Danish children. Int J Antimicrob Agents. 2017;49:573–8. https://doi.org/10.1016/j.ijantimicag.2017.01.011.
Mirsepasi H, Persson S, Struve C, Andersen LOB, Petersen AM, Krogfelt KA. Microbial diversity in fecal samples depends on DNA extraction method: EasyMag DNA extraction compared to QIAamp DNA stool mini kit extraction. BMC Res Notes. 2014;7:50.
Nakaune R, Nakano M. Efficient methods for sample processing and cDNA synthesis by RT-PCR for the detection of grapevine viruses and viroids. J Virol Methods. 2006;134:244–9.
Smith B, Li N, Andersen AS, Slotved HC, Krogfelt KA. Optimising bacterial DNA extraction from faecal samples: comparison of three methods. Open Microbiol J. 2011;5:14–7. https://doi.org/10.2174/1874285801105010014.
Neefs JM, De Wachter R. A proposal for the secondary structure of a variable area of eukaryotic small ribosomal subunit RNA involving the existence of a pseudoknot. Nucleic Acids Res. 1990;18:5695–704.
Yu Y, Lee C, Kim J, Hwang S. Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol Bioeng. 2005;89:670–9.
Sundberg C, Al-Soud WA, Larsson M, Alm E, Yekta SS, Svensson BH, et al. 454 pyrosequencing analyses of bacterial and archaeal richness in 21 full-scale biogas digesters. FEMS Microbiol Ecol. 2013;85:612–26. https://doi.org/10.1111/1574-6941.12148.
Takai K, Horikoshi K. Rapid Detection and Quantification of Members of the Archaeal Community by Quantitative PCR Using Fluorogenic Probes. Appl Environ Microbiol. 2000;66(11):5066–72.
Mortensen MS, Brejnrod AD, Roggenbuck M, Abu Al-Soud W, Balle C, Krogfelt KA, et al. The developing hypopharyngeal microbiota in early life. Microbiome. 2016;4:70. https://doi.org/10.1186/s40168-016-0215-9.
Kimer N, Pedersen JS, Tavenier J, Christensen JE, Busk TM, Hobolth L, et al. Rifaximin has minor effects on bacterial composition, inflammation and bacterial translocation in cirrhosis; a randomized trial. J Gastroenterol Hepatol. 2018;33(1):307–14. https://doi.org/10.1111/jgh.13852.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1. https://doi.org/10.1093/bioinformatics/btq461.
Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–8. https://doi.org/10.1038/nmeth.2604.
Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, et al. Chimeric 16S rRNA sequence formation and detection in sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011;21:494–504. https://doi.org/10.1101/gr.112730.110.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–41. https://doi.org/10.1128/AEM.01541-09.
Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010;26:266–7. https://doi.org/10.1093/bioinformatics/btp636.
Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26:1641–50. https://doi.org/10.1093/molbev/msp077.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. https://doi.org/10.1038/nmeth.f.303.
McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6:610–8. https://doi.org/10.1038/ismej.2011.139.
R Core Team. R: a language and environment for statistical. Computing. 2015; http://www.r-project.org/.
McMurdie PJ, Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217. https://doi.org/10.1371/journal.pone.0061217.
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, et al. Vegan: community ecology package. 2015. http://cran.r-project.org/package=vegan.
Thorsen J, Brejnrod A, Mortensen M, Rasmussen MA, Stokholm J, Al-Soud WA, et al. Large-scale benchmarking reveals false discoveries and count transformation sensitivity in 16S rRNA gene amplicon data analysis methods used in microbiome studies. Microbiome. 2016;4(1):62.
Russel J, Thorsen J, Brejnrod AD, Bisgaard H, Sørensen SJ, Burmølle M. DAtest: a framework for choosing differential abundance or expression method bioRxiv 2018. doi:https://doi.org/10.1101/241802.
Wickham H. ggplot2: Elegant Graphics for Data Analysis. 2nd ed. New York: Springer-Verlag; 2016. http://ggplot2.org
Acknowledgements
We express our deepest gratitude to the children and families that have participated in this cohort for all their support and commitment. We thank, Steffen Jørgensen, Bent Bjørn Roldgaard, Hengameh Mirsepasi-Lauridsen, and Susanne Schjørring for laboratory assistance in collection of the sample material and for updating the database.
Author contributions
This project was conceived and designed by KAK and AMP. BHJ, DR and BUA were responsible for contact to the families, collection of samples and questionnaires. LO’B performed DNA extractions. MSM, JW and ADB performed sequencing and bioinformatics analysis. MSM performed the microbiota analysis. Data interpretation were performed by MSM, BHJ, SJS, CRS, AMP, and KAK helped interpret the data. MSM and BHJ are the main author of this paper. All the authors have read, revised, and approved the final manuscript.
Funding
This work was partially supported by The Danish Council for Strategic Research, Innovation and Higher Education [grant: 2101-07-0023] granted to Professor Karen Angeliki Krogfelt. The funders had no role in the design of the study, collection, analysis, and interpretation of data, decision to publish, or preparation of the manuscript.
Availability of data and materials
The fastq files for all samples in this study are available in the Sequence Read Archive (BioProject ID: PRJNA491825. https://www.ncbi.nlm.nih.gov/bioproject/PRJNA491825). OTU tables, bioinformatics and R-scripts are available at http://mibi.galaxy.bio.ku.dk/martin/MortensenBMCMicrobiomeManuscript/
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was conducted in accordance with the Declaration of Helsinki and was approved by The National Committee on Health Research Ethics, protocol number (H-A-2008-111). Written informed consent was obtained from the parents or legal guardians of the participating children before enrolment. Only the children of parents capable of understanding oral and written Danish were included in the study. The study was approved by the Danish Data Protection Agency (protocol number 2013-41-2338).
Consent for publication
Written consent for publication has been obtained from the parents or legal guardians of all participants.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Rarefaction analysis of the species richness. Rarefaction curves showed a trend of an increasing diversity over time. Rarefaction curves were calculated for both observed richness and Shannon diversity index, grouped by time-point and with bars indicating standard deviations. The observed richness did not reach saturation before 20,000 reads, whereas the H′ reaches a maximum after 1000 reads (XLSX 20 kb)
Additional file 2:
Figure S2. Abundance of Core microbiota in each sample, by sample number, grouped by child. Barplot showing the abundance of the core microbiota in each sample. The samples are grouped by which child they are from and ordered in chronological order. (PDF 41 kb)
Additional file 3:
Table S1. Statistical output of all PERMANOVA analysis. Table S2. Table of bacteria, at all taxonomic levels, differential abundant with regard to exposures. Only taxa with an unadjusted p-value below 0.05 are included. Table S3. Table of genera being significantly correlated with both observed richness and Shannon diversity index. (PDF 70 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mortensen, M.S., Hebbelstrup Jensen, B., Williams, J. et al. Stability and resilience of the intestinal microbiota in children in daycare – a 12 month cohort study. BMC Microbiol 18, 223 (2018). https://doi.org/10.1186/s12866-018-1367-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-018-1367-5