
Abstract
Background
Activation of inflammasome contributes to the clearance of intracellular bacteria. C-terminus of E. coli EscI protein can activate NLRC4 (NLR family, CARD domain containing-4) inflammasome in macrophages. The purpose of this study was to determine if activation of NLRC4 inflammasome by EscI can reduce the colonization of Salmonella in mice.
Results
A recombinant S. typhimurium strain expressing fusion protein of the N-terminal SspH2 (a Salmonella type III secretion system 2 effector) and C-terminal EscI was constructed and designated as X4550(pYA3334-SspH2-EscI). In vitro assay showed that X4550(pYA3334-SspH2-EscI) significantly enhanced IL-1β and IL-18 secretion (P < 0.05) and pyroptotic cell death of mouse peritoneal macrophages, compared with those infected with control strain, X4550(pYA3334-SspH2). In vivo studies showed that colonization of X4550(pYA3334-SspH2-EscI) in both spleen and liver were significantly lower than that of X4550(pYA3334-SspH2) (P < 0.05). The bacterial counts of X4550(pYA3334-SspH2-EscI) in mice decreased, while those of X4550(pYA3334-SspH2) increased over the time after infection. Additionally, X4550(pYA3334-SspH2-EscI) induced a less pathological alteration in spleen and liver than X4550(pYA3334-SspH2).
Conclusion
Fusion protein SspH2-EscI may be translocated into macrophages and activate NLRC4 inflammasome, which limits Salmonella colonization in spleen and liver of mice.
Similar content being viewed by others


Background
Innate immune system plays a primary role in the rapid elimination of invading microorganisms, which occurs via the recognition of microbial pathogen-associated molecular patterns by the cellular pattern recognition receptors [1, 2]. Intracellular nucleotide binding domain leucine-rich repeat-containing receptor (NLR) can recognize microbial components that are transported to the cytoplasm through the bacterial secretion system, which can then activate inflammasome signalling [3, 4]. During this process, pro-caspase-1 is synthesized by activated macrophages and enriched in the inflammasome, ultimately being cleaved into the activated caspase-1 [5]. Activation of caspase-1 subsequently triggers IL-1β/IL-18 maturation and macrophage pyroptotic death. This pathway is important to defend against the colonization of intracellular bacteria in the intestinal tract and systemic circulation [6,7,8].
After infection, Salmonella can selectively secret cytoplasmic effectors through its type III secretion system (T3SS) [9,10,11]. These effectors regulate the host cells’ defense mechanisms to ensure the survival of the invading bacteria [12, 13]. Infection of Salmonella can result in both decreased breeding potential and increased fatality in host organism. Therefore, a key defensive step to protect against Salmonella infection is to reduce bacterial intracellular survival. Currently, there is no effective approach to induce adaptive immunity during the early stages of a Salmonella infection, thus it is critical to focus on innate immunity.
During the early stage of infection, Salmonella T3SS1 effectors are expressed to mediate bacterial infection. Once Salmonella enters host cells, T3SS2 effectors are expressed in order to mediate bacterial intracellular survival [14]. Though many proteins of Salmonella can activate intracellular inflammasome response, over the course of evolution, Salmonella has developed the ability to escape the inflammasome responses. This can occur through the T3SS1 protein PrgJ that can activate the NLRC4 inflammasome in macrophages, but is only expressed during the early stage of infection. When Salmonella successfully survives intracellularly, it no longer express PrgJ [15]. This suggests that if Salmonella strain can persistently express and transport PrgJ to the cytoplasm of host cells, it can enhance the activation of inflammasome and thereby inhibit the intracellular survival of bacteria [16]. It has also been reported that the immunization of the Listeria monocytogenesis strain that can enhance caspase-1 activation can confer protective immunity against a subsequent wild-type challenge [17]. Thus, it has been hypothesized that the Salmonella strain with the ability to enhance caspase-1 activation can strengthen the cell’s defense against Salmonella infection [18].
Previous reports have suggested that inflammasome activation mechanism can be used in the design of recombinant vaccines to limit the colonization of intracellular bacteria in vivo [19]. As previously reported, the N-terminus signal peptide of the Salmonella effector SspH2 can be recognized by T3SS2 and transported into the cytoplasm [20, 21]. The C-terminus of E. coli EscI protein can activate the NLRC4 (NLR family, CARD domain containing-4) inflammasome in macrophages [15]. In the present study, a recombinant Salmonella fusion expressing the N-terminus of Salmonella SspH2 and the C-terminus of E. coli EscI was constructed. The recombinant strain was tested for its ability to activate inflammasome and colonize in vivo in mouse.
Methods
Animals, plasmids and bacteria
Six-week-old female C57BL/6 mice were obtained from the Comparative Medicine Center of Yangzhou University (Yangzhou, China). This study was carried out in accordance with the regulations established by the Chinese Ministry of Science and Technology. The animal experiment protocol was approved by the Committee on the Ethics of Animal Experiments of Yangzhou University (Permit Number: 2007–0005). All surgery was performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering.
Plasmids pMD20 T (Amp+) and pYA3334 (asd+), E. coli DH5α (RˉMˉ, Ampˉ) and X6212 (asdˉ, NA+, RˉM+), attenuated S. Typhimurium strains X3730 (GalEˉ, Hsdˉ, Asdˉ, NA+, RˉM+) and X4550 (△crp-1, △cya-1, asdˉ, NA+, RˉM+) were used in this study as previously described [22, 23]. S. Enteritidis C50041 and E. coli O:157 were used for the amplification of sspH2 and escI genes, respectively. Bacterial strains were grown in Luria broth (LB) medium.
Construction of recombinant plasmid and S. typhimurium expressing SspH2-EscI fusion protein
The genomic DNA of bacteria C50041 and O:157 were extracted using the high pure PCR template preparation kit (Takara, Dalian, China) according to the manufacturer’s instructions. The nucleotide sequences of primers for polymerase chain reaction (PCR) were shown in Table 1, with the underlined segments indicating the restriction sites. The 5’-terminal sequence (1–453 bp) of the sspH2 gene was amplified from the C50041 strain using the primers SspH2-F1 (forward primer) and SspH2-R1 (reverse primer). The 3’-terminal sequence (205–426 bp) of the escI gene was amplified from O:157 strain using the primers EscI-F1 (forward primer) and EscI-R1 (reverse primer). The above two purified PCR products were then mixed for the overlap PCR splicing using the primers SspH2-F1 and EscI-R1. All PCR products were subsequently identified via agarose gel electrophoresis. The purified PCR product sspH2-escI (729 bp) was cloned into the plasmid pMD20 T and the recombinant plasmid was then transformed into E.coli DH5α for amplification. The recombinant plasmid was verified by restriction digestion and DNA sequencing. After digestion with Nco I and Sal I (Takara), the sspH2-escI gene was cloned into the plasmid pYA3334. The recombinant plasmid was named as pYA3334-SspH2-EscI and transformed into E.coli X6212. The recombinant plasmid pYA3334-SspH2-EscI was verified by enzyme digestion and DNA sequencing. The plasmid pYA3334-SspH2-EscI was then transformed into S. typhimurium X3730 for methylation modification. Finally, the modified plasmid pYA3334-SspH2-EscI was transformed into S. typhimurium X4550. The recombinant bacteria were designated as X4550(pYA3334-SspH2-EscI).
The purified PCR product sspH2 amplified from the C50041 strain using the primers SspH2-F1 (forward primer) and SspH2-R4 (reverse primer) was cloned into the plasmid pYA3334. The recombinant plasmid was named as pYA3334-SspH2 and the corresponding recombinant bacteria was named as X4550(pYA3334-SspH2).
The plasmid pYA3334 was used as a negative control and the corresponding recombinant bacteria was named as X4550(pYA3334).
Growth curve of recombinant S. typhimurium strains
The growth characteristic of recombinant bacteria was performed as previously described [23]. Briefly, single colony of recombinant bacteria was inoculated in LB medium. After being cultured with shaking at 37 °C overnight, 50 μl of bacteria was inoculated in 5 ml LB medium and cultured with shaking at 37 °C. OD600 was measured at different time to obtain the growth curve.
In vitro infection of mouse peritoneal macrophages
Peritoneal cells were collected by lavaging the mouse peritoneal cavity using RPMI 1640 culture medium supplemented with 10% fetal bovine serum (FBS) (Gibco, Carlsbad, CA). After washing with phosphate buffer saline (PBS), cells were suspended in RPMI 1640 complete medium (RPMI 1640 containing 10% FBS), seeded on 96-well plates, and cultured at 37 °C in 5% CO2 for 3 h.
Non-adherent cells were removed and cell density was adjusted to 20,000 cells per well. The adherent cells were pre-stimulated with 1 μg/ml E. coli lipopolysaccharide (LPS) (Sigma-Aldrich) to induce the expression of pro-IL-1β. The freshly cultured X4550(pYA3334-SspH2-EscI), X4550(pYA3334-SspH2) and X4550(pYA3334) were centrifuged at 1500 × g for 10 min and washed with PBS. The bacteria were resuspended in RPMI 1640 complete medium and added to the LPS-stimulated cells to the desired multiplicity of infection (MOI = 10, 50 and 100, respectively). The cell plate was centrifuged at 500 × g for 10 min to enhance the contact of bacteria with the cells. Infected cells were incubated at 37 °C for 30 min. The supernatants were then removed and washed with RPMI 1640 complete medium. Subsequently, RPMI 1640 complete medium containing 100 U/ml penicillin, 100 μg/ml streptomycin, and 1 μg/ml LPS were added to the cells (100 μl/well) to kill the extracellular bacteria. Cells were remained in culture at 37 °C and 5% CO2 for four hours [24, 25]. In all experiments, uninfected cells were used as controls. The cell morphology was observed using TS100-F inverted microscope (Nikon, Japan).
After culturing, the supernatants were collected and centrifuged at 2000 × g for 5 min to remove all dead bacteria and debris. Quantification of IL-1β and IL-18 was performed using cytometric bead array system (CBA) mouse IL-1β Flex Set and enzyme-linked immunosorbent assay kit (Biosciences, PharMingen, San Diego, CA) according to the manufacturer’s instructions. The flow cytometry was performed using FACSAria flow cytometer with FACSDiva software (Becton-Dickinson Immunocytometry Systems, BDIS, San Jose, CA).
The lactate dehydrogenase (LDH) release was measured using the cytotoxicity detection kit (Roche, Switzerland) according to the manufacturer’s instructions. The relative amount of released LDH was calculated as follows: %released LDH (sample) = (sample − medium background)/(total LDH − medium background) × 100% [26].
Cell plates were washed with PBS and the cells were collected for counting. Then the cells were lysed with lysing solution containing 1 mM phenylmethanesulfonyl fluoride (Westang, Shanghai) according to the manufacturer’s instructions. The intracellular bacteria were counted by coating on the LB agar plate containing NA (20 μg/ml) for culturing.
The intracellular caspase-1 activation were determined by FLICATM caspase-1 detection kit (Immunochemistry Technoligies Inc., Bloomington, MN) using flow cytometry according to the manufacturer’s instructions.
In vivo infection of mice
The freshly cultured bacteria were centrifuged at 1500 × g for 10 min and washed with PBS. Six-week-old C57BL/6 mice were intravenously injected with X4550(pYA3334), X4550(pYA3334-SspH2) and X4550(pYA3334-SspH2-EscI), respectively. Each mouse was infected with 1× 106 cfu using 100 μl PBS as vehicle. The mice intravenously injected with equivalent PBS were used as controls.
At different time points post-infection, the spleen and liver of mice were harvested to determine bacterial colonization. Briefly, the weight and size of the tissues were recorded. After grinding in 5 ml PBS, the suspension of spleen and liver tissues were ten-fold diluted in PBS and 100 μl suspension were then evenly plated on the LB agar containing nalidixic acid (NA, 20 μg/ml) for CFU enumeration.
Three weeks after infection, the pathological section of spleen and liver of mice were examined using hematoxylin-eosin (HE) staining.
Quantification of IL-6 and TNF-α were performed using CBA mouse inflammation kit (Biosciences, PharMingen, San Diego, CA) according to the manufacturer’s instructions.
Statistical analysis
Within each experiment, three to four replicate assays were conducted for each treatment and the average value was calculated for final statistical comparisons. All statistical analyses were performed by t-tests using SPSS software (Version 13.0 for Windows, Chicago, IL). A value of P ≤ 0.05 was considered to be statistically significant.
Results
Construction of recombinant plasmid and growth curve of recombinant bacteria
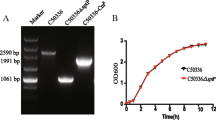
The PCR products of 5’-terminus of sspH2 gene (478 bp) and the 3’-terminus of escI gene (277 bp) were identified (Fig. 1a). The overlapping PCR product sspH2-escI gene was 729 bp as expected. Transformants could grow on LB agar plate without diaminopimelic acid (DAP) (Sigma). The recombinant plasmids subjected to restriction digestion with Sal I and Noc I were shown (Fig.1b, c). The sequencing results demonstrated that the recombinant plasmid pYA3334-SspH2 and pYA3334-SspH2-EscI was constructed successfully.

Construction of recombinant bacteria. a PCR products. M, Marker; 1, 5’-terminal of sspH2; 2, 3’-terminal of escI. b Recombinant plasmid pMD20 T-SspH2-EscI digested with Nco I and Sal I. M1-M2, Marker; 1, pMD20 T-SspH2-EscI. c Recombinant plasmid pYA3334-SspH2-EscI digested with Nco I and Sal I. M1-M2, Marker; 1, pYA3334-SspH2-EscI. d Growing curve of recombinant bacteria
The growth states of recombinant bacteria X4550(pYA3334-SspH2-EscI), X4550(pYA3334-SspH2) and X4550(pYA3334) were similar, suggesting that metabolism of the bacteria was not affected by transforming recombinant plasmid into S. typhimurium strain (Fig. 1d).
Pyroptotic cell death of mouse peritoneal macrophages after in vitro infection
IL-1β and IL-18 content in the supernatant of peritoneal macrophages following 4 h of infection (MOI = 50 or 100) was significantly higher than those in the unfected control (P < 0.05). Notably, infection with X4550(pYA3334-SspH2-EscI) induced significantly more IL-1β and IL-18 secretion from peritoneal macrophages than that induced by X4550(pYA3334-SspH2) or X4550(pYA3334) (P < 0.05, Fig. 2a).

In vitro infection of mouse peritoneal macrophages. C57BL/6 mouse peritoneal macrophages seeded on 96-well plates were pre-stimulated with 1 μg/ml E. coli lipopolysaccharide to induce the expression of pro-IL-1β for 3 h. The freshly cultured X4550(pYA3334-SspH2-EscI), X4550(pYA3334-SspH2) and X4550(pYA3334) were then added with the desired multiplicity of infection (MOI). The cell plate was centrifuged to enhance the contact of bacteria with the cells and the infected cells were then incubated for 30 min. The supernatants containing uninfected bacteria were replaced with RPMI 1640 complete medium (100 μl/well) containing 100 U/ml penicillin, 100 μg/ml streptomycin, and 1 μg/ml LPS prior to the start of the subsequent incubation. The uninfected cells were used as control. a Supernatant IL-1β and IL-18 levels at 4 h post-infection (hpi) with MOI 10, 50 and 100; b LDH release at 1, 3, 5 hpi with MOI 100 and 24 hpi with MOI 10, 50 and 100; c Intracellular caspase-1 activation at 1 h hpi with MOI 100; d Cell morphology at 24 hpi with MOI 100 (a, uninfection; b, infected with X4550(pYA3334); c, infected with X4550(pYA3334-SspH2); d, infected with X4550(pYA3334-SspH2-EscI), arrows show pyroptotic cell death); e The count of cells and intracellular bacteria at 24 hpi with MOI 100. The data shown are representative of three replicate experiments
LDH release assay indicated that X4550(pYA3334-SspH2-EscI) induced higher cytotoxicity than X4550(pYA3334-SspH2) and X4550(pYA3334) at 1, 3, 5 h post infection with MOI 100 and 24 h post infection with MOI 10, 50 and 100 (P < 0.05, Fig. 2b).
X4550(pYA3334-SspH2-EscI) induced higher level of caspase-1 activation than X4550(pYA3334-SspH2) and X4550(pYA3334) after infection. The result at 1 h post infection with MOI 100 was shown in Fig. 2c.
After 24 h, the morphology of X4550(pYA3334-SspH2-EscI)-infected cells (MOI = 100) was found to be markedly poor, and the integrity of the cell membrane was completely lost. Furthermore, the degree of injury induced by X4550(pYA3334-SspH2-EscI) was higher than that indeced by X4550(pYA3334-SspH2) and X4550(pYA3334) (Fig. 2d). Intracellular bacteria in the cells infected with X4550(pYA3334-SspH2-EscI) was significantly lower than that infected with X4550(pYA3334-SspH2) and X4550(pYA3334) (P < 0.05, Fig. 2e).
No significant difference was found between the cells infected with X4550(pYA3334-SspH2) and X4550(pYA3334) with regard to the IL-1β and IL-18 secretion, LDH release and intracellular bacterial counts.
Colonization of recombinant bacteria and pathology in mice
The spleen and liver of mice infected with X4550(pYA3334) or X4550(pYA3334-SspH2) had significant swelling with the infection time prolongation, when compared with those infected with X4550(pYA3334-SspH2-EscI). Six days after infection, the spleen size and weight of mice infected with X4550(pYA3334) or X4550(pYA3334-SspH2) were approximately four-fold greater than those infected with of X4550(pYA3334-SspH2-EscI). No significant differences were found between the mice infected with X4550(pYA3334-SspH2-EscI) and the uninfected controls (Fig. 3a).

In vivo infection of mice. Six-week-old C57BL/6 mice were intravenously injected with either freshly collected X4550(pYA3334), X4550(pYA3334-SspH2) or X4550(pYA3334-SspH2-EscI), 1× 106 cfu/mouse. Several days later, the weight of spleen (a), the bacterial colonization (b) in spleen and liver, and the contents of IL-6 and TNF in serum (c) were counted. Three weeks post-infection, the spleen and liver (d) of the mice were stained with hematoxylin-eosin for pathological assay, all scale bars represent 50 μm. Five mice were used in each treatment. The data shown are representative of three replicate experiments
One day after intravenous injection, the bacterial counts of X4550(pYA3334-SspH2-EscI) colonized in mice spleen and liver were significantly lower than those of X4550(pYA3334) and X4550(pYA3334-SspH2) (P < 0.05). As infection time extended, the bacteria counts in the spleen and liver of X4550(pYA3334-SspH2-EscI)-infected mice decreased significantly, with no bacteria being detected six days after infection. However, X4550(pYA3334)- and X4550(pYA3334-SspH2)- infected mice were observed to have a significant increase in bacterial counts over the time (Fig. 3b). No significant difference was found between X4550(pYA3334-SspH2) and X4550(pYA3334) with regards to the bacterial counts in the spleen and liver.
All infected mice could secrete IL-6 and TNF-α at 1 day post infection, while the uninfected mice secrete minimal level of IL-6 and TNF-α. Three days after infection, the IL-6 and TNF-α levels in serum of mice infected by X4550(pYA3334-SspH2-EscI) decreased, while those in mice infected with X4550(pYA3334-SspH2) and X4550(pYA3334) significantly increased (P < 0.05). No significant difference was found between X4550(pYA3334-SspH2) and X4550(pYA3334) (Fig. 3c).
Three weeks after infection, pathological analysis (Fig. 3d) showed stronger inflammatory responses in the spleen and liver of X4550(pYA3334)- and X4550(pYA3334-SspH2)- infected mice than those of X4550(pYA3334-SspH2-EscI)-infected mice and uninfected controls. Only a few small necrotic foci were found in the liver of X4550(pYA3334-SspH2-EscI)-infected mice, when compared with uninfected controls. In contrast, in the X4550(pYA3334)- and X4550(pYA3334-SspH2)- infected mice, large number of lymphocytes were observed in the splenic sinus, and many necrotic foci containing lymphocytes and necrotic hepatocytes were found in the liver. No significant pathological differences were found between X4550(pYA3334-SspH2) and X4550(pYA3334)-infected mice.
Discussion
Activation of NLR by microbial components can result in the subsequent activation of inflammasome in macrophages [1, 2] and is beneficial for the defense against intracellular bacteria [7, 8, 27, 28]. This is particularly important for the protection of intestinal mucosa and defense against systemic infection [6, 29]. Currently, the inflammasome mechanism has been predominantly stimulated with peptides in vitro [15]. However, due to the complex regulation by bacteria in the host cells, the responses of host cells against these peptides may be different from their response against the whole bacterium. Thus, it is more practical to study the inflammasome responses through bacterial infection, rather than peptide treatment. It has been reported that the recombinant Listeria monocytogenes that can enhance inflammasome response is attenuated and has a protective effect against virulent bacteria challenge [17]. Based on these previous findings, it has been suggested that an attenuated Salmonella vaccine candidate that enhances the inflammasome responses can also elevate cellular immunity against subsequent Salmonella infection [18]. To test this possibility, we sought to construct a recombinant Salmonella strain that can enhance inflammasome activation.
Salmonella pathogenicity islands (SPI)-1 and −2 express T3SS1 and T3SS2, respectively [30]. SPI-1 is mainly expressed in the intestines to promote the invasion of Salmonella into epithelial cells, while SPI-2 is mainly expressed in host cells to augment the survival of Salmonella in macrophages [14]. Reports have shown that NLRC4 can sense Salmonella proteins PrgJ and flagellin, which both contain a common C-terminal amino acid sequence [15, 26, 31]. However, over the course of its evolution, Salmonella has developed many evasion strategies to prevent NLRC4 detection in macrophages. For instance, SPI-1 and SPI-2 encode the rod proteins PrgJ and SsaI respectively, which form the needle in T3SS basal body [32]. NLRC4 can sense PrgJ, but not SsaI, due to one amino acid difference (V95) in the C-terminus between them [15]. Moreover, flagellin is repressed in the intracellular environment while SPI-2 T3SS is active [16, 33]. Taken together, it has been hypothesized that recombinant Salmonella expressing flagellin or PrgJ from a SPI-2 co-regulated promotor can be persistently detected via NLRC4 and completely cleared in vivo [16]. As reported, Salmonella effector SspH2 can be translocated by T3SS2 and colocalize with the polymerizing actin cytoskeleton [20]. The recombinant Salmonella expressing fusion protein of SspH2 and exogenous antigen can translocate the latter into the cytoplasm of macrophages [20, 21, 34, 35]. Furthermore, the SspH2 N-terminal amino acid sequence is conserved among different Salmonella strains and can be used as an efficient delivery vector [20]. EscI protein, the inner rod protein of enteropathogenic E. coli, is secreted in the early stage of infection [36, 37] and its C-terminal sequence can activate the NLRC4 inflammasome [15]. In this experiment, the N-terminus of SspH2 and the C-terminus of EscI were selected to construct the recombinant Salmonella expressing fusion protein SspH2-EscI.
Salmonella lacking asd gene has an obligatory requirement for DAP because the asd mutant will undergo lysis in environments deprived of DAP. The asd + plasmid containing the wild-type asd gene can complement the mutants to become a stable balanced-lethal system and be used to express exogenous antigens [38]. The △crp △cya Salmonella strain X4550 is avirulent and immunogenic in mice, and introduction of asd + plasmid pYA3334 into X4550 could completely restore avirulent [22]. It is reported that X4550 still can survive in mice for a long time [23]. Therefore, X4550 is usually used to express exogenous antigen to promote immunity without any antibiotic selection. In this experiment, X4550 was selected as the vector to express and transport fusion protein SspH2-EscI.
The intracellular caspase-1 activation and secretion of IL-1β and IL-18 are essential for inflammasome response in macrophages [7]. The recombinant bacteria expressing SspH2-EscI could significantly promote the secretion of IL-1β and IL-18 and the pyroptotic cell death of macrophages in in vitro infection when compared with bacteria expressing SspH2 only, suggesting that the intracellular recombinant Salmonella can successfully express fusion protein SspH2-EscI and the SspH2 N-terminus can be used as a signal to deliver EscI C-terminus into the host cells, resulting in activation of the NLRC4 inflammasome. Furthermore, in in vivo infection, the expression of SspH2-EscI, but not SspH2 alone, could inhibit the colonization of recombinant bacteria, suggesting that reduction of bacterial colonization in mice may be due to the activation of NLRC4 inflammasome by EscI in the cytoplasm.
So far, there are different interpretations about how the inflammasome pathway is used during immune defense [39]. Pyroptosis can lyse the host cells and the pathogen can then be phagocytosed by neutrophils, resulting in bacterial death. As reported, a sifA gene-mutated Salmonella can destroy the Salmonella-containing vacuole due to caspase-11 activation, but not due to the secretion of IL-1β and IL-18. It is also reported that the clearance of Burkholderia occurs, in part, due to the secretion of IL-1β and IL-18 after nasal infection. This indicates that pyroptosis is a defense mechanism to clear intracellular bacteria [40]. The anti-infection defense of recombinant L. monocytogenes that enhanced the inflammasome activation is due to caspase-1-induced pyroptosis, but not due to the secretion of IL-1β and IL-18 [17]. In this experiment, the inhibition of Salmonella colonization in mice several days after intravenous infection may be due to the pyroptosis observed in the earlier stages of infection. The definite mechanism should be further studied in the future using NLRC4¯ mice.
The inflammasome pathway was first named and characterized in 2002 [41] and has since seen great effort to elucidate its mechanism of action. This work can also lead to some applications, especially in the attenuated vaccine design. Because activated inflammasome is not specific to a particular bacteria, this will provide a general platform for the development of vaccine of not only Salmonella, but also other intracellular pathogens.
Conclusions
Through construction of recombinant Salmonella, we found that the expression of SspH2-EscI could enhance the activation of inflammasome responses in macrophages and decrease the colonization of bacteria in mice. We speculate that the fusion protein SspH2-EscI may be transported into the cytoplasm of macrophages and then activate NLRC4 inflammasome, which limits the colonization of Salmonella.
Abbreviations
- CBA:
-
Cytometric bead array system
- FBS:
-
Fetal bovine serum
- HE:
-
Hematoxylin-eosin
- LB:
-
Luria broth
- LDH:
-
Lactate dehydrogenase
- LPS:
-
Lipopolysaccharide
- NA:
-
Nalidixic acid
- NLR:
-
Nucleotide binding domain leucine-rich repeat-containing receptor
- NLRC4:
-
NLR family, CARD domain containing-4
- PBS:
-
Phosphate buffer saline
- PCR:
-
Polymerase chain reaction
- SPI:
-
Salmonella pathogenicity islands
- T3SS:
-
Type III secretion system
References
Skeldon A, Saleh M. The inflammasomes: molecular effectors of host resistance against bacterial, viral, parasitic, and fungal infections. Front Microbiol. 2011;2:15.
Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs dictates inflammasome specificity. Nature. 2012;477(7366):592–5.
Monie TP, Bryant CE, Gay NJ. Activating immunity: lessons from the TLRs and NLRs. Trends Biochem Sci. 2009;34(11):553–61.
Khameneh HJ, Mortellaro A. NLRC4 gets out of control. Nat Genet. 2014;46(10):1048–9.
Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med. 2010;207(8):1745–55.
Carvalho FA, Nalbantoglu I, Aitken JD, Uchiyama R, Su Y, Doho GH, et al. Cytosolic flagellin receptor NLRC4 protects mice against mucosal and systemic challenges. Mucosal Immunol. 2012;5(3):288–98.
Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477(7366):596–600.
Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11(12):1136–42.
Stevens MP, Humphrey TJ, Maskell DJ. Molecular insights into farm animal and zoonotic Salmonella infections. Phil Trans R Soc B. 2009;364:2709–23.
Lara-Tejero M, Kato J, Wagner S, Liu X, Galán JE. A Sorting platform determines the order of protein secretion in bacterial type III systems. Science. 2011;331:1188–91.
Yu XJ, McGourty K, Liu M, Unsworth KE, Holden DW. pH sensing by intracellular Salmonella induces effector translocation. Science. 2010;328(5981):1040–43.
McGhie EJ, Brawn LC, Hume PJ, Humphreys D, Koronakis V. Salmonella takes control: effector-driven manipulation of the host. Curr Opin Microbiol. 2009;12(1):117–24.
Lahiri A, Lahiri A, Iyer N, Das P, Chakravortty D. Visiting the cell biology of Salmonella infection. Microbes Infect. 2010;12:809–18.
Miao EA, Warren SE. Innate immune detection of bacterial virulence factors via the NLRC4 inflammasome. J Clin Immunol. 2010;30(4):502–6.
Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A. 2010;107(7):3076–80.
Miao EA, Rajan JV. Salmonella and caspase-1: a complex interplay of detection and evasion. Front Microbiol. 2011;2:85.
Warren SE, Duong H, Mao DP, Armstrong A, Rajan J, Miao EA, et al. Generation of a Listeria vaccine strain by enhanced caspase-1 activation. Eur J Immunol. 2011;41(7):1934–40.
Kupz A, Guarda G, Gebhardt T, Sander LE, Short KR, Diavatopoulos DA, et al. NLRC4 inflammasomes in dendritic cells regulate noncognate effector function by memory CD8(+) T cells. Nat Immunol. 2012;13(2):162–9.
Xu X, Hensel M. Systematic analysis of the SsrAB virulon of Salmonella enterica. Infect Immun. 2010;78(1):49–58.
Panthel K, Meinel KM, Domenech VES, Retzbach H, Igwe EI, Hardt WD, et al. Salmonella pathogenicity island 2-mediated overexpression of chimeric SspH2 proteins for simultaneous induction of antigen-specific CD4 and CD8 T Cells. Infect Immun. 2005;73(1):334–41.
Medina C, Camacho EM, Flores A, Mesa-Pereira B, Santero E. Improved expression systems for regulated expression in Salmonella infecting eukaryotic cells. PLoS One. 2011;6(8):e23055.
Meng FP, Ding J, Yu ZC, Han QL, Guo CC, Liu N, et al. Oral attenuated Salmonella typhimurium vaccine against MG7-Ag mimotope of gastric cancer. World J Gastroenterol. 2005;11(12):1833–6.
Xu XG, Zhao HN, Zhang Q, Ding L, Li ZC, Li W, et al. Oral vaccination with attenuated Salmonella enterica serovar Typhimurium expressing Cap protein of PCV2 and its immunogenicity in mouse and swine models. Vet Microbiol. 2012;157:294–303.
Pelegrin P, Barroso-Gutierrez C, Surprenant A. P2X7 receptor differentially couples to distinct release pathways for IL-1β in mouse macrophage. J Immunol. 2008;180:7147–57.
Hoffmann C, Galle M, Dilling S, Kappeli R, Muller AJ, Songhet P, et al. In macrophages, caspase-1 activation by SopE and the type III secretion system-1 of S. Typhimurium can proceed in the absence of flagellin. PLoS One. 2010;5(8):e12477.
Lightfield KL, Persson J, Trinidad NJ, Brubaker SW, Kofoed EM, Sauer JD, et al. Differential requirements for NAIP5 in activation of the NLRC4 inflammasome. Infect Immun. 2011;79(4):1606–14.
Becker CE, O’Neill LAJ. Inflammasomes in inflammatory disorders: the role of TLRs and their interactions with NLRs. Semin Immunopathol. 2007;29:239–48.
Shaw MH, Reimer T, Kim YG, Nunez G. NOD-like receptors (NLRs): bona fide intracellular microbial sensors. Curr Opin Immunol. 2008;20(4):377–82.
Byrne BG, Dubuisson JF, Joshi AD, Persson JJ, Swanson MS. Inflammasome components coordinate autophagy and pyroptosis as macrophage responses to infection. MBio. 2013;4(1):e00620–12.
Figueira R, Holden DW. Functions of the Salmonella pathogenicity island 2 (SPI-2) type III secretion system effectors. Microbiology. 2012;158(Pt5):1147–61.
Pereira MSF, Marques GG, Deilama JE, Zamboni DS. The Nlrc4 inflammasome contributes to restriction of pulmonary infection by flagellated Legionella spp. that trigger pyroptosis. Front Microbiol. 2011;2:33.
Marlovits TC, Kubori T, Sukhan A, Thomas DR, Galan JE, Unger VM. Structural insights into the assembly of the type III secretion needle complex. Science. 2004;306(5698):1040–2.
Yang X, Thornburg T, Suo Z, Jun S, Robison A, Li J, et al. Flagella overexpression attenuates Salmonella pathogenesis. PLoS One. 2012;7(10):e46828.
Jones-Carson J, McCollister BD, Clambey ET, Vazquez-Torres A. Systemic CD8 T-cell memory response to a Salmonella pathogenicity island 2 effector is restricted to Salmonella enterica encountered in the gastrointestinal mucosa. Infect Immun. 2007;75(6):2708–16.
Miao EA, Miller SI. A conserved amino acid sequence directing intracellular type III secretion by Salmonella typhimurium. Proc Natl Acad Sci U S A. 2000;97(13):7539–44.
Lefebre MD, Galan JE. The inner rod protein controls substrate switching and needle length in a Salmonella type III secretion system. Proc Natl Acad Sci U S A. 2014;111(2):817–22.
Sal-Man N, Deng W, Finlay BB. EscI: a crucial component of the type III secretion system forms the inner rod structure in enteropathogenic Escherichia coli. Biochem J. 2012;442(1):119–25.
Zhao Z, Li M, Luo D, Xing L, Wu S, Duan Y, et al. Protection of mice from Brucella infection by immunization with attenuated Salmonella enterica serovar typhimurium expressing A L7/L12 and BLS fusion antigen of Brucella. Vaccine. 2009;27:5214–9.
Kong W, Brovold M, Koeneman BA, Clark-Curtiss J, Curtiss 3rd R. Turning self-destructing Salmonella into a universal DNA vaccine delivery platform. Proc Natl Acad Sci U S A. 2012;109(47):19414–9.
Aachoui Y, Sagulenko V, Miao EA, Stacey KJ. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr Opin Microbiol. 2013;16(3):319–26.
Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-1β. Mol Cell. 2002;10:417–26.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31320103907, 31372414, 31372415) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Availability of data and materials
The dataset supporting the conclusions of this article is included within the article.
Authors’ contributions
This study was designed by XJ, MH, ZP and GC. Data collection and statistical analysis were performed by MH, WG, QY and SG. WZ, WL and CM were managed subject infection studies. YW wrote the first draft and MH, XZ, ZP, GC and XJ contributed to the final manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This study was approved by the Committee on the Ethics of Animal Experiments of Yangzhou University (Permit Number: 2007–0005). This study does not involve the use of human data or tissue.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hu, M., Zhao, W., Gao, W. et al. Recombinant Salmonella expressing SspH2-EscI fusion protein limits its colonization in mice. BMC Immunol 18, 21 (2017). https://doi.org/10.1186/s12865-017-0203-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12865-017-0203-2