
Abstract
Background
Restrictions to gene flow, genetic drift, and divergent selection associated with different environments are significant drivers of genetic differentiation. The black tiger shrimp (Penaeus monodon), is widely distributed throughout the Indian and Pacific Oceans including along the western, northern and eastern coastline of Australia, where it is an important aquaculture and fishery species. Understanding the genetic structure and the influence of environmental factors leading to adaptive differences among populations of this species is important for farm genetic improvement programs and sustainable fisheries management.
Results
Based on 278 individuals obtained from seven geographically disparate Australian locations, 10,624 high-quality SNP loci were used to characterize genetic diversity, population structure, genetic connectivity, and adaptive divergence. Significant population structure and differentiation were revealed among wild populations (average FST = 0.001–0.107; p < 0.05). Eighty-nine putatively outlier SNPs were identified to be potentially associated with environmental variables by using both population differentiation (BayeScan and PCAdapt) and environmental association (redundancy analysis and latent factor mixed model) analysis methods. Clear population structure with similar spatial patterns were observed in both neutral and outlier markers with three genetically distinct groups identified (north Queensland, Northern Territory, and Western Australia). Redundancy, partial redundancy, and multiple regression on distance matrices analyses revealed that both geographical distance and environmental factors interact to generate the structure observed across Australian P. monodon populations.
Conclusion
This study provides new insights on genetic population structure of Australian P. monodon in the face of environmental changes, which can be used to advance sustainable fisheries management and aquaculture breeding programs.
Similar content being viewed by others
Background
Local adaptation can occur when geographically discrete environmental conditions impose selection pressure on resident populations of widespread species. Within heterogeneous populations, local adaptation can arise when strong environmental selection pressure outweighs the influence of genetic drift and gene flow towards homogeneity [1]. The marine environment provides sufficient opportunities for homogenizing gene flow through passive dispersal of eggs and larvae with ocean currents and adult mobility [2]. Resident populations of discrete marine environments can initially differ genetically at a few sites in their genomes (e.g., allele frequency differences [3];, or can change rapidly to adapt to a new environment due to intense selection pressure [1, 4]. Thus, understanding how environmental features shape the genetic structure of populations is crucial because it helps to determine how populations evolve, along with the extent and scale of local adaptation [5,6,7]. Recently, local adaptation of marine organisms to environmental conditions has been investigated in commercially important marine invertebrates, including greenlip abalone (Haliotis laevigata) [8], eastern oyster (Crassostrea virginica) [9], American lobster (Homarus americanus) [10], and New Zealand scallop (Pecten novaezelandiae) [11]. However, to date, the influence of marine environmental conditions on the wild-type genetic diversity and structure of Australian giant black tiger shrimp (Penaeus monodon; Fabricius, 1798), is unknown.
Single nucleotide polymorphisms (SNP) have been demonstrated to be the most prevalent type of polymorphism within investigated genomes [4]. Moreover, SNP are becoming the marker of choice in genetic diversity, population genetics, and seascape genomics studies [12]. Additionally, genetic techniques offer a variety of methods using SNP datasets to detect putatively adaptive loci and assess how environmental parameters influence the extent of genetic variation within and among populations [13,14,15,16,17,18]. The application of these analyses has been divided into two main approaches: population differentiation (PD) and environmental association (EA). More specifically, the PD approach addresses traditional population differentiation by estimating the inherent variance in pairwise genetic differentiation values (FST) of each locus across the genome without assuming the effect of environmental variables on outlier SNP generation [13, 19]. Alternatively, the EA approach addresses environmental influence on outlier SNP generation by identifying candidate adaptive loci that covary with environmental factors based on allele frequency [20, 21]. EA applications are especially promising to detect loci under putative selection, because they do not require phenotypic nor experimental data, can define the contribution of environmental variables to adaptive genetic variation, and detect weak multilocus responses to environmental conditions [20,21,22,23]. Given the unique advantages and disadvantages of each approach, unique combinations of PD and EA may help to reduce the number of false negatives and promote uncovering potential genomic footprints of selection [20]. For example, both PD and EA methods have been employed for the successful detection of putatively adaptive loci and assessment of environmental parameter influence on genetic variation within and among populations [7, 10, 20, 24].
Penaeus monodon is widely distributed throughout the Indo-Pacific and is found along the western, northern, and eastern coasts of Australia [25,26,27]. The life history of P. monodon comprises an offshore planktonic larval phase, estuarine juvenile and adolescent phases, and an inshore adult phase [27]. Larvae and post-larvae can display daily vertical migration behaviours in synchrony with tidal current [28, 29]. A tagging study by Gribble et al. [26] revealed that shrimp in the Trinity Inlet closure in north Queensland moved an average distance of 31 km north (5–100 km) over an average of 74 days (10–111 days). Forbes et al. [30] found evidence for high gene flow among P. monodon samples collected in Madagascar, Mozambique, and South Africa, despite geographic separations of up to 2000 km. Thus, P. monodon exhibits the capacity for moderate to high-level gene flow (i.e., genetic connectivity) across large geographic distances. In Australia, previous genetic work on P. monodon determined the existence of population structure using microsatellites, allozymes and mitochondrial DNA (mtDNA) markers [25, 31,32,33]. These studies suggested that there was no genetic differentiation between northern and eastern populations, but that P. monodon from Western Australia was a separate genetic stock with reduced a number of allelic due to colonisation by east coast P. monodon individuals. Additionally, another previous study of Indo-Pacific P. monodon presented that P. monodon from Northern Territory separated into a discrete cluster while Queensland and Western Australia grouped with Thailand, Palau, PNG, Taiwan, Philippines, and Vietnam (Bac Lieu and Can Tho) in one cluster [34].
The main objective of this study is to resolve the population genetic structure of P. monodon in Australia using a high-resolution genome-wide SNP approach and identify any genomic signatures of local adaptation among geographically and environmentally discrete populations. More specifically, this study aimed to (1) assess the levels of gene flow, genetic diversity, and population structure among geographically discrete populations of Australian P. monodon for evidence of local adaptation, and (2) predict which environmental factors are most likely to influence Australian P. monodon population structure (i.e., drive local adaption). The implications of P. monodon genetic structure and local adaptation for management and conservation of Australian populations are discussed.
Results
Genotyping and SNP quality control
A total of 126,511 unique SNPs were obtained for each P. monodon individual (n = 283; see Methods) by DArTseq™ sequencing (see Additional file 1). Initial quality control assessment of DArTseq™ data using dartQC removed 115,811 SNPs or 91.5% of loci (Table 1). The remaining 10,700 loci were then tested for LD and HWE, which removed 76 and 0 SNPs, respectively. Following genotype filtering, five individuals (two and three individuals from Gulf of Carpentaria and Nickol Bay populations, respectively) were removed due to high rates of missing data (> 40%). A final set of 10,624 SNPs for each retained P. monodon individual (n = 278) were subjected to further analyses (see Additional files 1 and 2).
Population genetic diversity
For the retained 10,624 SNPs, mean allelic richness values (AR) ranged from 1.61 ± 0.48 for Nickol Bay (Western Australia) population to 1.75 ± 0.34 for Tiwi Island population (Northern Territory) (Table 2). A similar pattern was observed for percentage of polymorphic loci (PPL), which ranged from 62 to 87% for Nickol Bay and Tiwi Island populations, respectively. Contrastingly, average private allelic richness (APR) and average MAF of polymorphic loci were slightly higher for Western Australia (0.03 ± 0.14 and 0.18) than for both northern Queensland (Bramston Beach, Etty Bay, and Townville; 0.004 ± 0.039 and 0.13) and Northern Territory (Gulf of Carpentaria, Joseph Bonaparte Gulf, and Tiwi Island; 0.03 ± 0.13 and 0.15) populations, respectively.
Average observed heterozygosity (HO) across all seven populations ranged from 0.13 ± 0.15 to 0.16 ± 0.2 and average expected heterozygosity (HE) ranged from 0.15 ± 0.16 to 0.17 ± 0.17 while average multi-locus heterozygosity (Av. MLH) within populations ranged from 0.13 ± 0.01 to 0.16 ± 0.04 (Table 2). Among populations, Western Australia displayed the highest Ho and Av. MLH values (0.16 ± 0.2 and 0.16 ± 0.04, respectively) whereas northern Queensland showed the lowest levels (0.13 ± 0.15 and 0.13 ± 0.01, respectively). Moreover, average HO values were lower compared to average HE values in all northern Queensland and Northern Territory populations with the exception of Western Australia where average HO and HE values were similar (Table 2). Average FIS values for both northern Queensland and Northern Territory sites were positive and ranged from 0.10 to 0.14, while average FIS value for Western Australia population was negative, but close to zero (− 0.04). Estimated NeLD varied across populations and ranged from 165.4 in Western Australia (95% CI = 159.9–171.2) to 24,121 in Bramston Beach (95% CI = 11,024 - ∞), with Western Australia and Etty Bay populations having the lowest NeLD across all seven populations (Table 2).
Population differentiation and genetic structure
DAPC and NETVIEW analyses (see Methods and Additional file 1) suggested regional structure among Australian P. monodon populations, with evidence for three clusters: a north Queensland group (Bramston Beach, Etty Bay, and Townville), a Northern Territory group (Gulf of Carpentaria, Joseph Bonaparte Gulf, and Tiwi Island), and Western Australia (Nickol Bay) (Fig. 1). The individual density distribution of the first retained discriminant function indicated separation of north Queensland shrimp from those in the Northern Territory and Western Australia groups (Fig. 1b). Fine-scale population network analysis using Netview R provided greater resolution at the individual level between populations and demonstrated the same three genetic clusters at k-NN = 60 (Fig. 1c and d).

Population structure of 278 individuals of Penaeus monodon samples using 10,624 SNPs (BB: Bramston Beach, EB: Etty Bay, Townsville: TSV, Gulf of Carpentaria: GC, Joseph Bonaparte Gulf: JBG, Tiwi Island: TIW, and Nickol Bay: NKB). a Discriminant Analysis of Principal Components (DAPC) scatterplot and b an individual density plot on the first discriminant function created through the R package ‘adegenet ‘; and c and d Population networks constructed using the Netview P v.1.0 pipeline at kNN = 30 and 60. Dots represent individuals, whereas colored ellipses correspond to sampling origin
Across all P. monodon populations pairwise FST values based on unbiased [35] ranged from 0.001 to 0.107, while pairwise standard genetic distances [36] similarly ranged from 0.002 to 0.028 (Table 3). All pairwise FST comparisons were significant (p ≤ 0.05) except for pairwise comparison between Townsville and Etty Bay populations in north Queensland. Not surprisingly, the most geographically separated sites (Nickol Bay in Western Australia and three north Queensland populations at approx. 3091 to 3137 km apart) exhibited the largest significant pairwise FST (0.107) and standard genetic distance (0.028) values (Table 4). Subsequent regression-based analysis also demonstrated a significant linear relationship between pairwise geographic distance and pairwise genetic distance among all sampled populations (r2 = 0.86, p = 0.006; see Additional file 3).
AMOVA analysis also grouped populations by geographical region: north Queensland (Bramston Beach, Etty Bay, and Townsville), Northern Territory (Gulf of Carpentaria, Joseph Bonaparte Gulf, and Tiwi Island), and Western Australia (Nickol Bay). Hierarchical population genetic analyses indicated majority of variation (95.6%) being explained among individuals within populations (p < 0.01; Table 4). Differences among geographically discrete populations accounted for only 4.2% of the total variance (p < 0.01), while 0.2% of the total variance (p < 0.01) was explained by variation among populations within each group.
Detection of outlier loci
Initial Bayescan v.2.1 and PCAdapt analyses (FDR = 0.01 for both; see Methods) detected 582 and 514 outlier SNPs, respectively, among the 10,624 SNPs retained for each individual (see Additional file 1). LFMM analysis (K = 3 and FDR = 0.05; see Methods) identified 1449 outlier SNPs that were significantly associated with at least one of seven environmental parameters tested. Full RDA model supported the role of environmental variables in shaping the distribution of SNP genotypes (p < 0.01; R2 = 0.048; adjusted R2 = 0.031) with 43.5 and 30.2% of inherent genetic variation explained by the first and second RDA axes, respectively. Based on these two significantly constrained axes the RDA model collectively yielded 963 candidate outlier SNPs. Lastly, the rigorous evaluation of both PD and two EA analyses (see Methods) identified 425 and 161 outlier SNPs respectively, of which 89 (21 and 55%) overlapped (see Fig. 2 and Additional file 4), leaving 10,535 SNPs as neutral (see Additional file 5). Both outlier and neutral SNP were subjected to further analyses (see Additional file 1).

Venn diagram showing the total number of putative SNP significantly associated with at least one environmental variable. The total number of SNP is reported in each panel. The 89 outlier SNPs on the intersections (shaded area) were retained for further analysis
Genetic structure based on outlier and neutral markers
PCA analysis of all individuals for both neutral and outlier SNP separated Australian populations into the same three clusters as DAPC and Netview analyses (Western Australia separate from north Queensland and Northern Territory populations; see above). Considering the 10,535 neutral SNPs, PC1 and PC2 explained 24.8 and 14.7% of the total genetic variance, respectively (Fig. 3a). Considering the 89 outlier SNPs, PC1 and PC2 explained 5.8 and 1.0% of the total genetic variance, respectively (Fig. 3b).

Principal components analysis based allele frequencies for (a) 10,535 neutral SNPs loci and (b) 89 outlier SNPs loci (BB: Bramston Beach, EB: Etty Bay, Townsville: TSV, Gulf of Carpentaria: GC, Joseph Bonaparte Gulf: JBG, Tiwi Island: TIW, and Nickol Bay: NKB)
Pairwise FST estimates between populations differed depending on assessment of neutral or outlier loci (see Additional file 6). Genetic differentiation was significant (p < 0.05) for all pairwise comparisons when 10,535 neutral SNPs were assessed. Pairwise FST values ranged from 0.001 (Bramston Beach vs. Etty Bay and Joseph Bonaparte Gulf vs. Tiwi Island) to 0.1 (northern Queensland populations vs. Western Australia population). In a similar way, using 89 outlier loci, FST values between north Queensland populations and Western Australia (FST = 0.60–0.64) were in all cases higher than those between Northern Territory populations and Western Australia (FST = 0.19–0.22; p ≤ 0.01). FST values differed significantly between all populations within the Northern Territory (p ≤ 0. 04); however, no FST values differed significantly between populations within northern Queensland (p > 0.4).
Environmental factors association with genetic variation
ANOVA for RDA analyses demonstrated that five (Env_1, Env_2, Env_3, Env_5, and Env_7) and four (Env_2, Env_3, Env_5, Env_7) environmental factors (see Methods) were significantly associated with genetic variation in identified neutral and outlier SNP datasets (p = 0.001 and 0.001, adjusted R2 = 0.03 and 0.2), respectively (Fig. 4). When partitioning the relative importance of geographic proximity among sampled sites using partial RDA, the most important environmental predictors for neutral SNP were ranked as: Env_2 (F = 1.7, p = 0.001) > Env_1 (F = 1.5, p = 0.001) > Env_3 (F = 1.2, p = 0.001). For outlier SNP, Env_2 and Env_3 were both significant for putative adaptive genetic variation (p = 0.001); however, Env_2 explained 13% of inherent genetic variation while Env_3 explained 2.8%. When both neutral and outlier SNP datasets were combined, surface temperature max and min (Env_2 and Env_3 respectively) were the highest ranked predictors contributing to the genetic variation of Australian P. monodon. Variance partitioning based on neutral and outlier loci RDA models revealed that environmental effects explained 4.3 and 20.2% of genetic structure variation, while geographic location explained 12.9 and 2.8%, respectively. The remaining genetic structure variance was explained by the combination of environmental effect and geographic location.

Redundancy analysis on (a) 10,535 neutral loci and (b) 89 outlier SNPs allele frequencies in seven populations of Penaeus monodon in Australia. Explanatory variables (arrows) were RDA axes retained as important variable selection accounting for genetic variation (Env_2: surface temperature maximum, Env_3: surface temperature minimum, Env_5: surface phytoplankton mean, Env_7: benthic current velocity mean, and Env_1: surface salinity mean)
MRM correlation analysis conducted on geographic distance and pairwise population linearized FST values was significant (r2 = 0.9, p = 0.008). MRM models including surface temperature maximum (Env_2) and surface temperature minimum (Env_3) exhibited significant relationships with pairwise FST values (p = 0.001 and 0.008, adjusted R2 = 0.95 and 0.90), respectively (Table 5). Moreover, MRM models explained a large and significant proportion of genetic variability (> 90%), indicating that these environmental variables and geographic distance are significantly correlated and thus potentially constrain gene flow synergistically.
Gene ontology
Among the 89 SNPs that were found associated with environmental variables, 11 (12.4%) yielded matches to 173 total contigs in the P. monodon transcriptome with E value ≤1e-5 (see Additional file 7). Three of these 11 outlier SNPs exhibited BLAST hits in only one allele (ID: PM_10591, PM2065, and PM10621; see Additional file 7) and were thus excluded from translated protein comparison. Protein translation of the remaining eight outlier SNPs demonstrated the following: 1) five contained synonymous and non-synonymous mutations in two and four reading frames, 2) two contained synonymous and non-synonymous mutations in one and five reading frames, and 3) one contained synonymous and non-synonymous mutations in zero and six reading frames, respectively (see Additional file 8). However, only two of these eight outlier SNPs exhibited 100% pairwise identity with > 90% query cover to contigs within the P. monodon transcriptome (PM1958 and PM4714). More specifically, SNP PM1958 matched “calcium-activated chloride channel regulator”, “calcium-activated chloride channel regulator 2-like”, and “epithelial chloride channel-like” contigs while SNP PM4714 matched “uncharacterized protein APZ42_034504” or “Predicted: uncharacterized protein LOC108744461 isoform X4” contigs (Table 6). The remaining majority of outlier SNP (n = 78 or 87.6%) did not exhibit sufficiently strong matches to any contigs within the P. monodon transcriptome and were thus concluded to reside in non-coding (i.e., absent) genomic regions.
Discussion
The giant black tiger shrimp (P. monodon) is an important aquaculture species in Australia; however, to date only limited information is available regarding the influence of environmental pressures on the genetic structure of geographically discrete populations. Based on results from this study, three distinct genetic groups (i.e., stocks) were revealed across the geographic distribution range of Australian P. monodon: north Queensland (Bramston Beach, Etty Bay, and Townville), Northern Territory (Gulf of Carpentaria, Joseph Bonaparte Gulf, and Tiwi Island), and Western Australia (Nickol Bay). Of note is that the Western Australia population exhibited the lowest level of genetic variation of all assessed populations, which could be due to restricted gene flow between Western Australia and Northern Territory populations. Using multivariate analyses that considered both geographic distance and environmental factors it was determined that the majority of SNPs are neutral (n = 10,535), while a small subset of outliers are putatively adaptive (n = 89). Surface temperature maximum and minimum provided the strongest correlative explanation for the presence of these outliers (see below).
Population genetic diversity
Genetic diversity and stock assessments of Australian P. monodon were undertaken using all 10,624 SNP (i.e., neutral and outliers combined). Relative reduced genetic diversity (HO, HE, and Av.MLH) was observed for Australian P. monodon compared to other recently assessed crustacean species. More specifically, European green crab (Carcinus maenas) exhibited average HO and HE of 0.254 and 0.256 [38] while European lobster (Homarus gammarus) had HO and HE that ranged between 0.049–0.63 and 0.179–0.504, respectively [39] and scalloped spiny lobster (Panulirus homarus) had HO, HE, and Av.MLH that ranged between 0.166–0.184, 0.226–0.233, and 0.168–0.186, respectively [40]. The small reduction in genetic diversity observed for Australian P. monodon could be due to technical artefacts of the RADseq-based genotyping (i.e., null alleles) [41, 42] and/or sampling bias (i.e., Wahlund effect) [43, 44]. However, HO deficiency has also been observed in Bangladesh P. monodon using 10 microsatellite markers and 14 SNP loci [45], so the exact cause is yet to be fully resolved, but is more likely technical than biological.
In the Western Australia (Nickol Bay) population, heterozygosity (i.e., HO and HE) were equal or higher than other populations despite AR and PPL being slightly lower (0.01–0.02) (see Results). This slight reduction in AR and PPL is most likely due to the smaller sample size in our study for the Western Australia population [46]. SNP genotyping of this Western Australia population revealed equal or higher heterozygosity, despite previous allozyme and microsatellite based demonstrations of reduced heterozygosity and number of alleles in P. monodon from the same Western Australia region [31, 47]. These previous P. monodon population genetics studies concluded that observed reductions in heterozygosity and number of alleles were most likely driven by founder effects or bottleneck events. As such, based on the entire Western Australia SNP dataset encompassing 10,624 genome-wide SNPs, Western Australia P. monodon appears from our dataset to not have undergone a bottleneck (i.e., heterozygosity not reduced), but rather has retained similar heterozygosity to Northern Territory and north Queensland populations. Differences between our dataset and those using microsatellites, allozymes and mtDNA may simply be due to the level of genetic resolution of these markers when comparing allelic diversity, where our high-resolution sampling captured more of the true genetic diversity within the P. monodon genome. Likewise, Lemopoulos et al. [48] demonstrated that SNPs were more informative than microsatellites for applications that required individual-level genotype information (i.e., estimating relatedness and genetic diversity with low sample sizes from small populations). North Queensland and Northern Territory P. monodon populations exhibited FIS values > 10%, which exceeded a general FIS threshold of ~ 10% suggested by Moss et al. [49] in a study of the effects of inbreeding on survival and growth of Pacific white shrimp (Litopenaeus vannamei). However, slightly positive FIS values observed here are most likely a result of DArTSeq genotyping technical artifacts (ie., null alleles) rather than of biological origin. Furthermore, the large NeLD estimates determined for all Australian P. monodon populations are likely to reduce any possible effect of inbreeding [50]. It is also noteworthy, that FIS values in this study are lower than observed in wild Pacific Ocean L. vannamei collected from Panama to Mexico (FIS = 0.53) [51] and along the Mexican coast (FIS = 0.36) [52].
Population differentiation and genetic structure
Pairwise FST analysis for north Queensland and Northern Territory Australian P. monodon populations revealed relatively weak genetic structuring (0–0.039), except for Western Australia (0.069–0.107), which had the highest levels detected (Table 3). Additionally, genetic differentiation increased proportionally with geographic distance and followed the IBD pattern across the entire sampled range, which is in agreement with previous observations based on mtDNA and microsatellite sequences [25, 31, 33]. However, this finding contrasts the FST values observed between Western Australia and Northern Territory (0.116) and Queensland (0.032) [34]. As such, the significantly different genetic composition between Western Australia and all other populations may reflect the effects of restricted gene flow and genetic drift, which is not surprising for P. monodon given the relatively short offshore planktonic larval phase (approximately 20 days), during which larvae and post-larvae disperse and migrate to nursery areas (e.g., in-shore areas and mangrove estuaries) [27]. Moreover, the evidence for adaptive divergence among Australian P. monodon populations presented here (see below) suggests that divergent selection may be contributing to genetic divergence despite genetic drift [53]. Regardless of the exact cause, these results suggest that there is a restriction in gene flow between geographically disparate Australian P. monodon populations.
Assessment of population structure using FST, DAPC and Netview analyses revealed regional structure in Australian P. monodon with three major population groupings: north Queensland (Bramston Beach, Etty Bay, and Townsville), Northern Territory (Gulf of Carpentaria, Joseph Bonaparte Gulf, and Tiwi Island), and Western Australia (Nickol Bay) (Fig. 1). One possible explanation for this genetic structure is the presence of biogeographic barriers between eastern, northern, and Western Australia that caused restricted gene flow between P. monodon populations. Upwelling of deep cold water along the north-western Australian coastline, which has existed since the Late Miocene period, is one biogeographical barrier known to prevent gene flow between Western Australia and other Australian regions [54]. Moreover, repeated declines in sea surface levels of 100–140 m during the Pleistocene caused biogeographic isolation between northern and eastern Australia due to repeated emergence of land bridges between northern Australia, Torres Strait, and New Guinea [55,56,57,58]. Similar patterns in genetic structure between Australian populations have been observed for other marine species such as Australian Spanish mackerel (Scomberomorus commerson) [59], mud crab (Scylla serrata) [60], brown tiger prawn (Penaeus esculentus) [61], and barramundi (Lates calcarifer) [62]. Previous P. monodon genetic differentiation investigations based on allozyme, mitochondrial DNA and microsatellite markers also revealed significant genetic partitioning among Australian and south-east Asian P. monodon populations indicating significant bio-geographic barriers to dispersal [25, 31, 33, 34]. Accordingly, the current observed genetic differentiation among Australian P. monodon populations appear to be driven by the presence of biogeographic barriers between eastern, northern, and Western Australia that effectively limited or prevented gene flow over evolutionary timescales.
Evidence for local adaptation
PD and EA analyses collectively identified 89 overlapping outlier SNPs, which accounted for 21 and 55% of total outlier SNPs detected, respectively (Fig. 2). This observation is in agreement with previous studies that also found EA approaches to perform better than PD approaches for detection of loci under putative divergent selection [10, 20]. Moreover, the observed levels of genetic structure based on these 89 outlier SNPs agreed with the genetic structure observed when all 10,535 neutral SNPs were considered.
Analysis of population structure based on 89 outlier SNPs using PCA presented a similar spatial pattern as to PCA based on 10,535 neutral SNPs in the form of three groups: north Queensland, Northern Territory, and Western Australia (Nickol Bay). Several causes may contribute to the same population structure being determined when either neutral or outlier loci were used (e.g., life history, natural selection, and environmental heterogeneity [63,64,65]; however, determination of the exact cause requires further investigation. Our findings agree with previous SNP-based studies conducted on other species with a planktonic larval phase (e.g., Eulachon (Thaleichthys pacificus), European green crab (C. maenas), and Atlantic salmon (Salmo salar)) that were determined to have low level gene flow and genome-wide divergence [38, 64, 66], respectively. These outlier-based analyses suggest that key environmental factors (see Results) and geographic distance, synergistically or independently, contributed to the generation of the 89 outlier SNPs observed among Australian P. monodon populations (i.e., adaptive divergence drivers). This conclusion is supported by other studies that also demonstrated that association between environmental variables and outlier SNPs could be indicative of local adaptation [9, 10, 23, 67, 68].
Environmental variables contributing to genetic structure
RAD, pRAD and MRM analyses demonstrated a strong relationship between geographic distance, environmental distance, and genetic differentiation in both neutral (n = 10,535) and outlier (n = 89) SNPs inherent to wild Australian P. monodon. This significant positive correlation suggests that: 1) gene flow between sampled sites may be impacted by natural selection or 2) geographically discrete Australian P. monodon populations have adapted to region-specific environmental variables (e.g., temperature). Geographic isolation due to biogeographic barriers (see above) could have caused initial level of genetic differentiation between P. monodon populations with subsequent divergent selection imposed by environmental factors leading to increased differentiation across evolutionary time. This interpretation is supported by RDA analysis, which indicated that the effects of surface temperature maximum and minimum (Env_2 and Env_3 respectively) were more pronounced than geographic distance for both neutral and outlier SNP.
RAD and partial RAD analyses conducted on outlier SNP demonstrated that surface temperature maximum (Env_2) explained five and six times more genetic variation than was explained by surface temperature minimum (Env_3), respectively. Therefore, it is plausible that surface temperature maximum (Env_2) could be the predominant driver of local adaption among Australian P. monodon populations. In a review of local adaptation among populations of marine invertebrates, Sanford and Kelly [69] found that approximately 44% of surveyed studies provided evidence of local adaptation to temperature. Moreover, several studies targeting marine invertebrates demonstrated the role of sea surface temperature in shaping genetic differences among populations (e.g., American lobster (H. americanus) [10], European green crab (C. maenas) [68], eastern oyster (C. virginica) [9], sea scallop (Placopecten magellanicus) [67], and giant California sea cucumber (Parastichopus californicus) [23]). Winter sea surface temperature was also demonstrated to be the most likely driver of local adaptation and limiter of gene flow among North American populations of C. maenas [68]. In Australia, sea surface temperatures have become significantly warmer between 1950 and 2007 along north-eastern and north-western tropical coasts by 0.12 °C and 0.11 °C per decade, respectively [70], while sea surface temperature in south-western Australia has risen by 0.026 to 0.034 °C per year between 1985 and 2004 [71]. Moreover, surface temperature maximum and minimums occur during summer and winter, respectively, which is when P. monodon broodstock emigrate out of estuaries into foreshores for spawning and, thus, extreme swings could influence individual fitness (i.e., breeding success). Further studies are needed to elucidate the existence and extent of population-specific thermal adaptation among Australia P. monodon populations and the potential functional genomics implications of the identified 89 outlier SNPs.
Highlights from gene ontology
The combination of PD and EA approaches identified 89 outlier SNPs that are potential targets for local adaptation within the Australian P. monodon genome. Only 11 of these 89 outlier SNPs matched P. monodon transcriptome contigs [72] with subsequent protein translation of eight outlier SNPs demonstrating more non-synonymous changes than synonymous changes across all six reading frames. Despite the greater likelihood that non-synonymous mutations will have functional implications [73, 74], all 11 of these outlier SNPs with P. monodon transcriptome matches should be considered as candidates for future research into divergent selection driven local adaptation among these geographically discrete populations [75].
Of these 11 outlier SNPs, one matched a contig annotated as “calcium-activated chloride channel regulator” (CLCA) (Table 6). CLCA is involved in cellular physiology functions such as neuronal and cardiac action, muscle contraction, and epithelial secretion [76,77,78]. In Pacific white shrimp L. vannamei, a similar calcium-activated chloride channel gene was shown to be expressed in gill cells and exhibit potential involvement in osmoregulation because of observed response to salinity challenge [79]. As such, future P. monodon gene-by-environment studies should consider investigating the potential role of this gene in local adaptation to population-specific environmental conditions.
The remaining 78 outlier SNPs are presumed to be located in non-coding regions absent from the transcriptome, or in genes with low-level expression that were not captured within initial transcriptome assembly [80,81,82].
Implications for aquaculture management and future research directions
The black tiger shrimp is an important aquaculture and fishery species in Australia. Therefore, the identification of genetic stock units among wild populations is crucial for fishery and aquaculture broodstock management. Neutral and adaptive population structure findings suggested that Australian P. monodon should be managed as three separate stocks: north Queensland, Northern Territory, and Western Australia. The levels of genetic diversity revealed in the present study using both neutral and outlier SNP are useful for aquaculture purposes such as selective breeding programs, maintaining stock diversity, and distinguishing hatchery stocks from wild populations. Moreover, in addition to reduced gene flow, thermal adaptation is also likely to contribute to a greater divergence in the Western Australia population. Regardless, the strong genetic structure and presence of rare alleles in all seven P. monodon populations suggests that these populations should be managed accordingly to maintain genetic integrity. Environmental factors can influence genetic diversity and population structure in marine species and an accurate understanding of this influence can both improve fisheries management and help predict responses to environmental change [67]. This study suggests that both geographical distance and environmental factors interact to influence the genetic structure of Australian P. monodon; however, the magnitude of influence for each of these factors is hard to determine conclusively. This study also provides evidence for environmentally-driven selection pressure on geographically discrete populations, which can be utilized to help ensure sustainable management of Australian P. monodon (e.g., guidance for future re-establishment of populations inhabiting similar thermal gradients). Practically, a population that is potentially under local adaptive pressures may be an important source of private or rare alleles that can enhance population resistance to future environmental change (e.g., naturally or via selective breeding programs) or assist natural migration [83, 84].
Conclusions
We utilized a SNP dataset containing 10,624 loci to determine genetic population structure and local adaptation across seven populations of Australian black tiger prawn P. monodon (n = 278 individuals). This study provides novel insights that assist the development and implementation of P. monodon aquaculture and fishery management practices within Australia. Analysis of population structure using both neutral (n = 10,535) and outlier (n = 89) SNPs suggest that Australian P. monodon should be managed as three separate stocks (north Queensland, Northern Territory, and Western Australia) and that geographically discrete P. monodon populations have likely undergone local adaptation to region-specific thermal regimes. Future studies should investigate the role that outlier SNPs potentially play in local adaptation in order to advance wild stock structure preservation and help facilitate selective breeding programs.
Methods
Sample collection and DNA extraction
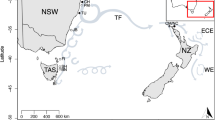
Individual P. monodon (n = 283) were collected from seven locations around Australia between 2015 and 2017 (Fig. 5). More specifically, wild adult P. monodon were obtained by trawling from Bramston Beach (n = 60, BB), Etty Bay (n = 50, EB), Townsville (n = 22, TSV), Joseph Bonaparte Gulf (n = 34, JBG), Tiwi Island (n = 56, TIW), Gulf of Carpentaria (n = 35, GC), and Nickol Bay (n = 26, NKB) (see Additional file 9). Pleopod tissue samples were collected from each adult individual using pre-sterilised scissors followed by preservation in 80% ethanol and − 20 °C storage until DNA extraction.

Map showing the seven localities where 283 wild Penaeus monodon samples were collected (BB: Bramston Beach, EB: Etty Bay, Townsville: TSV, Gulf of Carpentaria: GC, Joseph Bonaparte Gulf: JBG, Tiwi Island: TIW, and Nickol Bay: NKB). The map was created using R package ‘ggplot2’ version 3.2.1 (https://github.com/tidyverse/ggplot2) [85] and R package ‘ozmaps’ version 0.3.6 (https://github.com/mdsumner/ozmaps) [86]
Genomic DNA (gDNA) was extracted from each pleopod tissue sample following a modified cetyl trimethyl ammonium bromide (CTAB) protocol [44, 87]. Briefly, 100 mg preserved tissue was digested in a CTAB buffer with 20 mg/mL proteinase K for ≥6 h at 55 °C under gentle agitation followed by one phenol–chloroform–isoamyl alcohol phase separation, two chloroform – isoamyl alcohol phase separations, overnight isopropanol precipitation (4 °C), two 70% ethanol washes, and elution in Tris-EDTA buffer [88]. All gDNA extractions were subsequently cleaned using Sephadex™ G-50 [89]. Extracted gDNA samples were assessed for quantity and quality using Nanodrop 1000 Spectrophotometer (260:230 nm and 260:280 nm ratios; ThermoFisher Scientific Pty Ltd., Australia) and 0.8% agarose gel containing GelGreen (ThermoFisher Scientific Pty Ltd., Australia). All gDNA samples were then diluted with Tris-EDTA buffer to a normalized final concentration of 50 ng/μl before shipping to Diversity Arrays Technology Sequencing (DArTseq™; Canberra Australia) for library preparation and DArTseq™ sequencing.
SNP genotyping and quality control
SNP discovery and genotyping were performed by Diversity Arrays Technology using DArTseq™ hybridization-based sequencing technology on next-generation sequencing (NGS) platforms as per [87, 90, 91]. DArTSeq™ sequences were further filtered using custom dartQC python scripts (available at https://github.com/esteinig/dartqc). More specifically, SNP sequences matching the following criteria were removed from the dataset (see Additional file 1): (1) average read depth < 7, (2) average repeatability < 90%, (3) call rate < 80%, (4) similar sequence clusters > 0.95, and (5) minor allele frequency (MAF) < 0.02.
All individuals and remaining loci were subsequently tested for missing data and linkage disequilibrium (LD) using PLINK 1.9 [92]. All individuals with high rate of missing data (> 40%) were excluded from the dataset. From the pairs of loci determined to be in LD (with a Pearson coefficient of determination (r2) threshold of 0.2), SNPs with the lowest call rate value were removed. Lastly, remaining SNP dataset for each population was then assessed for deviation from Hardy–Weinberg equilibrium (HWE) using R package dartR [93] and all SNP that significantly deviated from HWE (p < 0.0001) were removed.
Genetic diversity, population differentiation and structure
Several measures were used to estimate population genetic variation and differentiation (see Additional file 1). Observed heterozygosity (HO), expected heterozygosity (HE), and inbreeding coefficient (FIS) were calculated using the divBasic function of R package diveRsity version 1.9.90 [94]. FIS values were determined using 95% confidence intervals and 1000 bootstrap replicates. Allelic richness (AR), private allelic richness (ARA), and percentage of polymorphic loci (PPL) were calculated for each SNP using rarefaction to avoid sampling size bias in HP-RARE version 1.1 [95]. Multilocus heterozygosity (MLH) was then calculated for all individuals from each population using R package inbreedR version 0.3.2 [96]). Effective population size (NeLD) was also estimated for each population using NeEstimator version 2.1 based on LD and random mating model [97].
Genetic differences among populations and individuals were calculated using R package StAMPP version 1.5.1 [37]. Pairwise genetic differentiation values (FST), along with confidence intervals and probability (p) values between populations, were calculated according to Wright [98] and updated by Weir and Cockerham [35]. Nei [36] standard genetic distances (Ds) were calculated across loci in a pairwise comparison between populations and individuals. To evaluate hierarchical population genetic differentiation, an analysis of molecular variance (AMOVA) was implemented using ARLEQUIN version 3.5.2.2 [18]. Statistical significance of each variance component was assessed using 1000 permutations for each of the following hierarchical comparisons: 1) among groups, 2) among populations within groups, and 3) within populations. To determine isolation by distance (IBD), a Mantel test was conducted for the full SNP dataset (10,624) using the mantel.randtest function in R package ade4 [99] with 999 permutations.
To determine the number of genetic groups a Discriminant Analysis of Principal Components (DAPC) was used on individual genotypes in R package adegenet version 2.1.1, with number of possible clusters (cluster values (K) of 1–20) determined by running 60 iterations of ‘find.clusters’ function [100]. Bayesian information criterion (BIC) values, which allow choosing the optimal K value, were averaged across all 60 iterations and standard deviation estimated for each K value. Additionally, genotypic relationships between individuals with no prior population assumptions were assessed using R package NetView [101, 102]. NetView was run through multiple K-nearest neighbour (kNN) values between 1 and 60 to assess both broad and fine scale population structure.
Environmental data collection and processing
Environmental factors considered ecologically relevant to P. monodon [26, 27] were selected from the Bio-ORACLE database (http://www.bio-oracle.org/downloads-to-email.php) [103, 104]. QGIS version 2.18.11 (https://www.qgis.org/) was used to extract values of 23 marine environmental variables (see Additional file 10) based on latitude and longitude of all seven sampling locations around northern Australia (see Fig. 1; Additional file 9). Marine data layers were then produced using monthly averages of climate data from 2000 to 2014 for all seven sampling locations (Fig. 1). Correlation estimates generated for marine data layers and filtered SNP datasets for each population using Pearson Test (R software cor and cor.test functions) identified seven marine environmental variables with Pearson coefficient of correlation (r) between − 0.75 and 0.75. Marine environmental variables retained for further analysis were: 1) surface salinity mean (Env_1), 2) surface temperature maximum (Env_2), 3) surface temperature minimum (Env_3), 4) surface current velocity mean (Env_4), 5) surface phytoplankton mean (Env_5), 6) benthic temperature mean (Env_6), and 7) benthic current velocity mean (Env_7) (see Additional file 7).
Detection of loci under selection
The possible effects of divergent selection on the overall pattern of genetic differentiation was assessed by combining the results of two independent population differentiation (PD) and two independent environmental association (EA) approaches (see Additional file 1). First, BayeScan version 2.1 [14, 105] and R package PCAdapt [13] were used to identify loci under selection (i.e., two independent PD approaches). Second, to identify environmental variables associated with genetic variation, redundancy analysis (RDA) in the R package vegan version 2.5–2 [106, 107] and latent factor mixed models (LFMM) in the R package LEA version 2.0.0 [16] were explored (i.e., two independent EA approaches). All four approaches were run on the same SNP dataset using strict parameters (see below). Only candidate outlier SNP identified by all four approaches were retained for subsequent analyses (see below).
BayeScan version 2.1 [14, 105] was run with 20 pilot runs of 5000 iterations followed by 100,000 iterations and an additional burn-in of 50,000 iterations. Alpha levels and FST values were ordered from largest to smallest and candidate loci were determined using false discovery rate (FDR) control levels of 0.001, 0.005, 0.01, 0.05, and 0.1 using Bayescan 2.01 function plot_R.r [108]. The R package PCAdapt [13] with K = 2 and min.maf = 0.05 was used to detect outlier loci based on principal component analysis (PCA) by assuming that markers excessively related with population structure are candidates for local adaptation. The candidate loci were determined using FDR control levels ranging from 0.01 to 0.1.
The linear model redundancy analysis (RDA) was run in R package vegan version 2.5.2 [106]. The vegan function ordistep, which applies a stepwise per mutational ordination method, was used to perform the ‘optimal’ model that corresponded to the highest adjusted coefficient of determination (adj R2). To test the significance of the final RDA model, the vegan function anova.cca was run with 999 permutations then outlier SNPs were identified on each of the first two constrained axes (p = 0.001) using the function outliers. Finally, LFMM models were run using R package LEA version 2.0.0 [16] for each environmental variable with a total number of 10,000 iterations and a burn-in of 5000 iterations. The specified result (K = 3) corresponded with the number of population clusters identified in DAPC from five repetitions. Z-scores for each locus were combined across five replicates and FDR were evaluated using FDR-adjusted p values. Candidate loci were determined for each marine environmental variable (n = 7; see above) and all candidate loci with FDR-adjusted p ≤ 0.05 were retained for subsequent analyses (see below).
Environmental factors association with genetic variation
To examine adaptive and neutral patterns of variation within SNP obtained across all seven collection locations (Fig. 1), PCA was performed on the retained environmental outlier (n = 89) and neutral (n = 10,535) loci using R package adegenet version 2.1.1 [100]. Retained outlier and neutral loci were then used to calculate genetic differences among populations and individuals using R package StAMPP version 1.5.1 [37].
To examine the relative contribution of geographical distances and environmental variables and select which environmental factors best explain Australian P. monodon genetic structure, redundancy (RDA) and partial redundancy (pRDA) analyses were conducted for both retained outlier and neutral locus allele frequencies using R package vegan. Analyses of variance (ANOVA) with 999 permutations were used to assess the significance of each environmental parameter within the RDA for all RDA and partial pRDA tests. The vegan function ordistep with 999 permutations was used to evaluate each environmental variable and perform the ‘optimal’ model (i.e., model that best explained genetic structure of each discrete population).
To determine whether isolation by distance (IBD) could explain geographic patterns of differentiation in retained outlier loci (see above), multiple regression on distance matrices (MRM) [109] was conducted in R package ecodist [110] with 1000 permutations. Pairwise population FST values calculated in StAMPP and least-cost geographic distances calculated in marmap were used as response values and explanatory variables, respectively, and then MRM models were generated using the marine environmental variables that significantly correlated with genetic variation of retained outlier loci. Note that MRM models focused on the relationship between geographic distance and environmental variables to determine the significance of environment factors on gene flow among populations [68].
Gene ontology
Each outlier SNP was assessed against an assembled P. monodon transcriptome [72] using Geneious Prime version 2019.1.3 [111]. Contigs within assembled P. monodon transcriptome that exhibited strong pairwise similarity with any outlier SNP (E-value ≤1e− 5) were compiled and reported along with contig annotations provided by Huerlimann et al. [72]. All SNP that exhibited transcriptome matches were translated into protein sequence using all six reading frames (three and three in 5′ – 3′ and 3′ – 5′, respectively) to assess if mutation caused synonymous or non-synonymous change.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its Additional Files. The P. monodon transcriptome used for gene ontology in this study is available from the National Center for Biotechnology Information database under the following accession numbers (https://www.ncbi.nlm.nih.gov/nuccore/GGLH00000000.1): BioProject: PRJNA421400, BioSamples: SAMN08741487-SAMN08741521, SRA: SRP127068 (RR6868116-SRR6868172), TRA: GGLH00000000, (Huerlimann, et al. [72]).
Change history
20 July 2021
A Correction to this paper has been published: https://doi.org/10.1186/s12864-021-07794-w
Abbreviations
- AMOVA:
-
Analysis of molecular variance
- BIC:
-
Bayesian information criterion
- IBD:
-
Isolation by distance
- DAPC:
-
Discriminant Analysis of Principal Components
- SNP:
-
Single nucleotide polymorphisms
- PD:
-
Population differentiation
- EA:
-
Environmental association
- LD:
-
Linkage disequilibrium
- HWE:
-
Hardy–Weinberg equilibrium
- RDA:
-
Redundancy analysis
- pRDA:
-
partial redundancy analysis
- LFMM:
-
Latent factor mixed models
- FDR:
-
False discovery rate
- MRM:
-
Multiple regression on distance matrices
- PCA:
-
Principal component analysis
- F ST :
-
Pairwise genetic differentiation values
- CLCA:
-
Calcium-activated chloride channel regulator
- NeLD :
-
Effective population size
- kNN:
-
Multiple K-nearest neighbor
References
Gandon S, Michalakis Y. Local adaptation, evolutionary potential and host–parasite coevolution: interactions between migration, mutation, population size and generation time. J Evol Biol. 2002;15(3):451–62.
Waples RS. Separating the wheat from the chaff: patterns of genetic differentiation in high gene flow species. J Hered. 1998;89(5):438–50.
Beaumont MA, Balding DJ. Identifying adaptive genetic divergence among populations from genome scans. Mol Ecol. 2004;13(4):969–80.
Morin PA, Luikart G, Wayne RK. SNPs in ecology, evolution and conservation. Trends Ecol Evol. 2004;19(4):208–16.
Hecht BC, Matala AP, Hess JE, Narum SR. Environmental adaptation in Chinook salmon (Oncorhynchus tshawytscha) throughout their north American range. Mol Ecol. 2015;24(22):5573–95.
Jump AS, Hunt JM, Marti'nez-izquierdo JA, Penuelas J. Natural selection and climate change: temperature-linked spatial and temporal trends in gene frequency in Fagus sylvatica. Mol Ecol. 2006;15(11):3469–80.
Rellstab C, Gugerli F, Eckert AJ, Hancock AM, Holderegger R. A practical guide to environmental association analysis in landscape genomics. Mol Ecol. 2015;24(17):4348–70.
Sandoval-Castillo J, Robinson NA, Hart AM, Strain LW, Beheregaray LB. Seascape genomics reveals adaptive divergence in a connected and commercially important mollusc, the greenlip abalone (Haliotis laevigata), along a longitudinal environmental gradient. Mol Ecol. 2018;27(7):1603–20.
Bernatchez S, Xuereb A, Laporte M, Benestan L, Steeves R, Laflamme M, Bernatchez L, Mallet MA: Seascape genomics of eastern oyster (Crassostrea virginica) along the Atlantic coast of Canada. Evol Appl. 2019;12(3):587–609.
Benestan L, Quinn BK, Maaroufi H, Laporte M, Clark FK, Greenwood SJ, Rochette R, Bernatchez L. Seascape genomics provides evidence for thermal adaptation and current-mediated population structure in American lobster (Homarus americanus). Mol Ecol. 2016;25(20):5073–92.
Silva CN, Gardner JP. Identifying environmental factors associated with the genetic structure of the New Zealand scallop: linking seascape genetics and ecophysiological tolerance. ICES J Mar Sci. 2015;73(7):1925–34.
Nadeem MA, Nawaz MA, Shahid MQ, Doğan Y, Comertpay G, Yıldız M, Hatipoğlu R, Ahmad F, Alsaleh A, Labhane N. DNA molecular markers in plant breeding: current status and recent advancements in genomic selection and genome editing. Biotechnol Biotechnol Equip. 2018;32(2):261–85.
Luu K, Bazin E, Blum MG. pcadapt: an R package to perform genome scans for selection based on principal component analysis. Mol Ecol Resour. 2017;17(1):67–77.
Foll M, Gaggiotti O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics. 2008;180(2):977–93.
Oksanen J, Blanchet F, Kindt R, Legendere P, Minchin P, O’Hara R. Vegan: Community Ecology Package. R Package Vegan, Version 2.2-1; 2015.
Frichot E, François O. LEA: an R package for landscape and ecological association studies. Methods Ecol Evol. 2015;6(8):925–9.
Whitlock MC, Lotterhos KE. Reliable detection of loci responsible for local adaptation: inference of a null model through trimming the distribution of FST. Am Nat. 2015;186(S1):S24–36.
Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and windows. Mol Ecol Resour. 2010;10(3):564–7.
Jensen JD, Foll M, Bernatchez L. The past, present and future of genomic scans for selection. Mol Ecol. 2016;25(1):1–4.
Dalongeville A, Benestan L, Mouillot D, Lobreaux S, Manel S. Combining six genome scan methods to detect candidate genes to salinity in the Mediterranean striped red mullet (Mullus surmuletus). BMC Genomics. 2018;19(1):217.
De Mita S, Thuillet AC, Gay L, Ahmadi N, Manel S, Ronfort J, Vigouroux Y. Detecting selection along environmental gradients: analysis of eight methods and their effectiveness for outbreeding and selfing populations. Mol Ecol. 2013;22(5):1383–99.
Hendricks S, Anderson EC, Antao T, Bernatchez L, Forester BR, Garner B, Hand BK, Hohenlohe PA, Kardos M, Koop B. Recent advances in conservation and population genomics data analysis. Evol Appl. 2018;11(8):1197–211.
Xuereb A, Kimber CM, Curtis JM, Bernatchez L, Fortin MJ. Putatively adaptive genetic variation in the giant California Sea cucumber (Parastichopus californicus) as revealed by environmental association analysis of restriction-site associated DNA sequencing data. Mol Ecol. 2018.
Andrew SC, Jensen H, Hagen IJ, Lundregan S, Griffith SC. Signatures of genetic adaptation to extremely varied Australian environments in introduced European house sparrows. Mol Ecol. 2018;27(22):4542–55.
Brooker A, Benzie J, Blair D, Versini J-J. Population structure of the giant tiger prawn Penaeus monodon in Australian waters, determined using microsatellite markers. Mar Biol. 2000;136(1):149–57.
Gribble N, Atfield J, Dredge M, White D, Kistle S. Sustainable Penaeus monodon (Black Tiger prawn) Populations for Broodstock Supply the Department of Primary Industries, Queensland. Brisbane: Queensland Dept. of Primary Industries; 2003.
Motoh H: Biology and ecology of Penaeus monodon. In: Proceedings of the First International Conference on the Culture of Penaeid Prawns/Shrimps, 4-7 December 1984, Iloilo City, Philippines: Aquaculture Department, Southeast Asian Fisheries Development Center; 1985. p. 27–36.
Rothlisberg PC. Vertical migration and its effect on dispersal of penaeid shrimp larvae in the Gulf of Carpentaria, Australia; 1982.
Queiroga H, Blanton J. Interactions between behaviour and physical forcing in the control of horizontal transport of decapod crustacean larvae. Adv Mar Biol. 2004;47:107–214.
Forbes A, Demetriades N, Benzie J, Ballment E: Allozyme frequencies indicate little geographic variation among stocks of giant tiger prawn Penaeus monodon in the south-west Indian Ocean. Afr J Mar Sci. 1999,21:271–7.
Benzie J, Frusher S, Ballment E. Geographical variation ion allozyme frequencies of populations of Penaeus monodon (Crustacea: Decapoda) in Australia. Mar Freshw Res. 1992;43(4):715–25.
Benzie J, Ballment E, Frusher S. Genetic structure of Penaeus monodon in Australia: concordant results from mtDNA and allozymes. Aquaculture. 1993;111(1–4):89–93.
Benzie J, Ballment E, Forbes A, Demetriades N, Sugama K, Moria S. Mitochondrial DNA variation in Indo-Pacific populations of the giant tiger prawn, Penaeus monodon. Mol Ecol. 2002;11(12):2553–69.
Waqairatu SS, Dierens L, Cowley JA, Dixon TJ, Johnson KN, Barnes AC, Li Y. Genetic analysis of black Tiger shrimp (Penaeus monodon) across its natural distribution range reveals more recent colonization of Fiji and other South Pacific islands. Ecol Evol. 2012;2(8):2057–71.
Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38(6):1358–70.
Nei M. Genetic distance between populations. Am Nat. 1972;106(949):283–92.
Pembleton LW, Cogan NO, Forster JW. StAMPP: an R package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Mol Ecol Resour. 2013;13(5):946–52.
Jeffery NW, DiBacco C, Van Wyngaarden M, Hamilton LC, Stanley RR, Bernier R, FitzGerald J, Matheson K, McKenzie C, Nadukkalam Ravindran P. RAD sequencing reveals genomewide divergence between independent invasions of the European green crab (Carcinus maenas) in the Northwest Atlantic. Ecol Evol. 2017;7(8):2513–24.
Jenkins TL, Ellis CD, Stevens JR: SNP discovery in European lobster (Homarus gammarus) using RAD sequencing. Conservation Genetics Resources 2019, 11(3):253–7.
Al-Breiki RD, Kjeldsen SR, Afzal H, Al Hinai MS, Zenger KR, Jerry DR, Al-Abri MA, Delghandi M. Genome-wide SNP analyses reveal high gene flow and signatures of local adaptation among the scalloped spiny lobster (Panulirus homarus) along the Omani coastline. BMC Genomics. 2018;19(1):690.
Andrews KR, Luikart G. Recent novel approaches for population genomics data analysis. Mol Ecol. 2014;23(7):1661–7.
Lal MM, Southgate PC, Jerry DR, Bosserelle C, Zenger KR. A parallel population genomic and hydrodynamic approach to fishery management of highly-dispersive marine invertebrates: the case of the Fijian black-lip pearl oyster Pinctada margaritifera. PLoS One. 2016;11(8):e0161390.
De Meeûs T. Revisiting F IS, F ST, Wahlund effects, and null alleles. J Hered. 2017;109(4):446–56.
Lal MM, Southgate PC, Jerry DR, Zenger KR. Genome-wide comparisons reveal evidence for a species complex in the black-lip pearl oyster Pinctada margaritifera (Bivalvia: Pteriidae). Sci Rep. 2018;8(1):191.
Alam MM, Pálsson S. Population structure of the giant tiger shrimp Penaeus monodon in Bangladesh based on variation in microsatellites and immune-related genes. Mar Biol Res. 2016;12(7):706–14.
Kalinowski ST. Counting alleles with rarefaction: private alleles and hierarchical sampling designs. Conserv Genet. 2004;5(4):539–43.
Benzie J. Population genetic structure in penaeid prawns. Aquac Res. 2000;31(1):95–119.
Lemopoulos A, Prokkola JM, Uusi-Heikkilä S, Vasemägi A, Huusko A, Hyvärinen P, Koljonen ML, Koskiniemi J, Vainikka A. Comparing RADseq and microsatellites for estimating genetic diversity and relatedness—implications for brown trout conservation. Ecol Evol. 2019;9(4):2106–20.
Moss DR, Arce SM, Otoshi CA, Doyle RW, Moss SM. Effects of inbreeding on survival and growth of Pacific white shrimp Penaeus (Litopenaeus) vannamei. Aquaculture. 2007;272:S30–7.
Frankham R, Ballou J, Briscoe D. Introduction to conservation genetics. Cambridge: Cambridge University Press; 2002. p. 617.
Valles-Jimenez R, Cruz P, Perez-Enriquez R. Population genetic structure of Pacific white shrimp (Litopenaeus vannamei) from Mexico to Panama: microsatellite DNA variation. Mar Biotechnol. 2004;6(5):475–84.
Vela Avitúa S, Montaldo HH, Márquez Valdelamar L, Campos Montes GR, Castillo Juárez H. Decline of genetic variability in a captive population of Pacific white shrimp Penaeus (Litopenaeus) vannamei using microsatellite and pedigree information. Electron J Biotechnol. 2013;16(4):9–9.
Funk WC, Lovich RE, Hohenlohe PA, Hofman CA, Morrison SA, Sillett TS, Ghalambor CK, Maldonado JE, Rick TC, Day MD. Adaptive divergence despite strong genetic drift: genomic analysis of the evolutionary mechanisms causing genetic differentiation in the island fox (Urocyon littoralis). Mol Ecol. 2016;25(10):2176–94.
Williams S, Benzie J. Evidence of a biogeographic break between populations of a high dispersal starfish: congruent regions within the indo-West Pacific defined by color morphs, mtDNA, and allozyme data. Evolution. 1998;52(1):87–99.
Galloway RW. Late cainozoic environments in Australia. Ecol Biogeography Austr. 1981:53–80.
Sloss CR, Nothdurft L, Hua Q, O’Connor SG, Moss PT, Rosendahl D, Petherick LM, Nanson RA, Mackenzie LL, Sternes A. Holocene Sea-level change and coastal landscape evolution in the southern Gulf of Carpentaria, Australia. Holocene. 2018;28(9):1411–30.
Chivas AR, Garcia A, van der Kaars S, Couapel MJ, Holt S, Reeves JM, Wheeler DJ, Switzer AD, Murray-Wallace CV, Banerjee D. Sea-level and environmental changes since the last interglacial in the Gulf of Carpentaria, Australia: an overview. Quat Int. 2001;83:19–46.
Voris HK. Maps of Pleistocene Sea levels in Southeast Asia: shorelines, river systems and time durations. J Biogeogr. 2000;27(5):1153–67.
Buckworth R, Newman S, Ovenden J, Lester R, McPherson G. The stock structure of northern and western Australian Spanish mackerel. Final Report. Fisheries Res Dev Corporation Proj. 1998;159:225.
Gopurenko D, Hughes JM. Regional patterns of genetic structure among Australian populations of the mud crab, Scylla serrata (Crustacea: Decapoda): evidence from mitochondrial DNA. Mar Freshw Res. 2002;53(5):849–57.
Ward RD, Ovenden JR, Meadows JR, Grewe PM, Lehnert SA. Population genetic structure of the brown tiger prawn, Penaeus esculentus, in tropical northern Australia. Mar Biol. 2006;148(3):599–607.
Loughnan SR, Smith-Keune C, Beheregaray LB, Robinson NA, Jerry DR. Population genetic structure of barramundi (Lates calcarifer) across the natural distribution range in Australia informs fishery management and aquaculture practices. Mar Freshwater Res. 2019.
Sexton JP, Hangartner SB, Hoffmann AA. Genetic isolation by environment or distance: which pattern of gene flow is most common? Evolution. 2014;68(1):1–15.
Moore JS, Bourret V, Dionne M, Bradbury I, O'Reilly P, Kent M, Chaput G, Bernatchez L. Conservation genomics of anadromous Atlantic salmon across its north American range: outlier loci identify the same patterns of population structure as neutral loci. Mol Ecol. 2014;23(23):5680–97.
Forester BR, Jones MR, Joost S, Landguth EL, Lasky JR. Detecting spatial genetic signatures of local adaptation in heterogeneous landscapes. Mol Ecol. 2016;25(1):104–20.
Candy JR, Campbell NR, Grinnell MH, Beacham TD, Larson WA, Narum SR. Population differentiation determined from putative neutral and divergent adaptive genetic markers in eulachon (Thaleichthys pacificus, Osmeridae), an anadromous Pacific smelt. Mol Ecol Resour. 2015;15(6):1421–34.
Van Wyngaarden M, Snelgrove PV, DiBacco C, Hamilton LC, Rodríguez-Ezpeleta N, Zhan L, Beiko RG, Bradbury IR. Oceanographic variation influences spatial genomic structure in the sea scallop, Placopecten magellanicus. Ecol Evol. 2018;8(5):2824–41.
Jeffery NW, Bradbury IR, Stanley RR, Wringe BF, Van Wyngaarden M, Lowen JB, McKenzie CH, Matheson K, Sargent PS, DiBacco C. Genomewide evidence of environmentally mediated secondary contact of European green crab (Carcinus maenas) lineages in eastern North America. Evol Appl. 2018.
Sanford E, Kelly MW. Local adaptation in marine invertebrates. Annu Rev Mar Sci. 2011;3:509–35.
Lough J. Shifting climate zones for Australia's tropical marine ecosystems. Geophys Res Lett. 2008;35(14).
Pearce A, Feng M. Observations of warming on the Western Australian continental shelf. Mar Freshw Res. 2007;58(10):914–20.
Huerlimann R, Wade NM, Gordon L, Montenegro JD, Goodall J, McWilliam S, Tinning M, Siemering K, Giardina E, Donovan D: De novo assembly, characterization, functional annotation and expression patterns of the black tiger shrimp (Penaeus monodon) transcriptome. Sci Rep. 2018;8(1):1–14.
Parmley JL, Chamary J, Hurst LD. Evidence for purifying selection against synonymous mutations in mammalian exonic splicing enhancers. Mol Biol Evol. 2005;23(2):301–9.
Lawrie DS, Messer PW, Hershberg R, Petrov DA. Strong purifying selection at synonymous sites in D. melanogaster. PLoS Genet. 2013;9(5):e1003527.
Lebeuf-Taylor E, McCloskey N, Bailey SF, Hinz A, Kassen R. The distribution of fitness effects among synonymous mutations in a gene under directional selection. eLife. 2019;8:e45952.
Hu D, Ansari D, Zhou Q, Sasor A, Hilmersson KS, Bauden M, Jiang Y, Andersson R. Calcium-activated chloride channel regulator 1 as a prognostic biomarker in pancreatic ductal adenocarcinoma. BMC Cancer. 2018;18(1):1096.
Nyström EE, Birchenough GM, van der Post S, Arike L, Gruber AD, Hansson GC, Johansson ME. Calcium-activated Chloride Channel regulator 1 (CLCA1) controls mucus expansion in Colon by Proteolytic activity. EBioMedicine. 2018.
Zhang S, Chen Y, An H, Liu H, Li J, Pang C, Ji Q, Zhan Y. A novel biophysical model on calcium and voltage dual dependent gating of calcium-activated chloride channel. J Theor Biol. 2014;355:229–35.
Hu D, Pan L, Zhao Q, Ren Q. Transcriptomic response to low salinity stress in gills of the Pacific white shrimp, Litopenaeus vannamei. Mar Genomics. 2015;24:297–304.
Harrisson KA, Amish SJ, Pavlova A, Narum SR, Telonis-Scott M, Rourke ML, Lyon J, Tonkin Z, Gilligan DM, Ingram BA. Signatures of polygenic adaptation associated with climate across the range of a threatened fish species with high genetic connectivity. Mol Ecol. 2017;26(22):6253–69.
Harrisson KA, Pavlova A, Telonis-Scott M, Sunnucks P. Using genomics to characterize evolutionary potential for conservation of wild populations. Evol Appl. 2014;7(9):1008–25.
Tigano A, Shultz AJ, Edwards SV, Robertson GJ, Friesen VL. Outlier analyses to test for local adaptation to breeding grounds in a migratory arctic seabird. Ecol Evol. 2017;7(7):2370–81.
Golbuu Y, Gouezo M, Kurihara H, Rehm L, Wolanski E. Long-term isolation and local adaptation in Palau’s Nikko Bay help corals thrive in acidic waters. Coral Reefs. 2016;35(3):909–18.
Ofori BY, Beaumont LJ, Stow AJ. Cunningham's skinks show low genetic connectivity and signatures of divergent selection across its distribution. Ecol Evol. 2017;7(1):48–57.
Wickham H, Chang W, Henry L, Pedersen T, Takahsahi K, Wilke C, Woo K, Yutani H. ggplot2: Elegant Graphics for Data Analysis (version 3.2. 1). New York: Springer-Verlag; 2016.
Sumner M, Cook D, Pebesma E. ozmaps: Australia Maps. R package version 0.3.6. In: R package version 0.3.6 edn; 2020. https://github.com/mdsumner/ozmaps.
Adamkewicz SL, Harasewych M. Systematics and biogeography of the genus Donax (Bivalvia: Donacidae) in eastern North America. Am Malacol Bull. 1996;13:97–104.
Gomes I, Prinz M, Pereira R, Meyers C, Mikulasovich RS, Amorim A, Carracedo A, Gusmão L. Genetic analysis of three US population groups using an X-chromosomal STR decaplex. Int J Legal Med. 2007;121(3):198–203.
GE HLS: Illustra AutoSeq G-50 and AutoSeq 96 dye terminator removal. In: Data files 28–9175-28. Illustra AutoSeq G-50 documents; 2007. p. 1–4.
Sansaloni CP, Petroli CD, Carling J, Hudson CJ, Steane DA, Myburg AA, Grattapaglia D, Vaillancourt RE, Kilian A. A high-density diversity arrays technology (DArT) microarray for genome-wide genotyping in eucalyptus. Plant Methods. 2010;6(1):16.
Kilian A, Wenzl P, Huttner E, Carling J, Xia L, Blois H, Caig V, Heller-Uszynska K, Jaccoud D, Hopper C. Diversity arrays technology: a generic genome profiling technology on open platforms. Data Prod Anal Popul Genomics Methods Protoc. 2012:67–89.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, De Bakker PI, Daly MJ. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75.
Gruber B, Unmack PJ, Berry OF, Georges A. dartr: An r package to facilitate analysis of SNP data generated from reduced representation genome sequencing. Mol Ecol Resour. 2017.
Keenan K, McGinnity P, Cross TF, Crozier WW, Prodöhl PA. diveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol. 2013;4(8):782–8.
Kalinowski ST. Hp-rare 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes. 2005;5(1):187–9.
Stoffel MA, Esser M, Kardos M, Humble E, Nichols H, David P, Hoffman JI. inbreedR: an R package for the analysis of inbreeding based on genetic markers. Methods Ecol Evol. 2016;7(11):1331–9.
Do C, Waples RS, Peel D, Macbeth G, Tillett BJ, Ovenden JR. NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (ne) from genetic data. Mol Ecol Resour. 2014;14(1):209–14.
Wright S. The genetical structure of populations. Ann Eugenics. 1949;15(1):323–54.
Dray S, Dufour A-B, Thioulouse J. Package ‘ade4’: analysis of ecological data: exploratory and Euclidean methods in environmental sciences; 2018.
Jombart T: adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24(11):1403–5.
Steinig EJ, Neuditschko M, Khatkar MS, Raadsma HW, Zenger KR. netview p: a network visualization tool to unravel complex population structure using genome-wide SNPs. Mol Ecol Resour. 2016;16(1):216–27.
Neuditschko M, Khatkar MS, Raadsma HW. NetView: a high-definition network-visualization approach to detect fine-scale population structures from genome-wide patterns of variation. PLoS One. 2012;7(10):e48375.
Assis J, Tyberghein L, Bosch S, Verbruggen H, Serrão EA, De Clerck O. Bio-ORACLE v2. 0: extending marine data layers for bioclimatic modelling. Glob Ecol Biogeogr. 2018;27(3):277–84.
Tyberghein L, Verbruggen H, Pauly K, Troupin C, Mineur F, De Clerck O. Bio-ORACLE: a global environmental dataset for marine species distribution modelling. Glob Ecol Biogeogr. 2012;21(2):272–81.
Foll M. Bayescan v2. 1 user manual. Ecology. 2012;20:1450–62.
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’hara R, Simpson GL, Solymos P, Stevens MHH, Wagner H. Package ‘vegan’. Commun Ecol Package Version. 2013;2(9).
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, R. B. O'Hara, Simpson GL, Solymos P et al: vegan: Community Ecology Package. R package version 2.5-2. 2018.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995:289–300.
Lichstein JW. Multiple regression on distance matrices: a multivariate spatial analysis tool. Plant Ecol. 2007;188(2):117–31.
Goslee SC, Urban DL. The ecodist package for dissimilarity-based analysis of ecological data. J Stat Softw. 2007;22(7):1–19.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–9.
Acknowledgements
We would like to thank the trawlermen for wild P. monodon sample collection; Julie Goldsbury for laboratory mentorship; Maria G. Nayfa, Eike J. Steinig, and Brooke L. Whitelaw for support during initial data analysis; Richard C. Edmunds for assistance with manuscript preparation; and Research Institute for Aquaculture No.1, Dinhbang, Tuson, Bacninh, Vietnam for supporting NTTV. All authors declare no conflict of interest.
Funding
This project was supported by The Australian Research Council Industrial Transformation Research Hub for Advanced Prawn Breeding (IH130200013). The first author, NTTV was supported by a Vietnam International Cooperation Department via Biotechnology of Aquaculture Scholarship and a James Cook University School/Faculty Scholarship. Funding bodies had no direct involvement in study design, sample collection, data analysis, data interpretation, or manuscript preparation.
Author information
Authors and Affiliations
Contributions
NTTV was responsible for all data analyses and drafting the manuscript. MJS facilitated the collection of wild samples. JLG, SRK, and CNSS participated in laboratory work, data analyses and manuscript editing. KRZ and DRJ supervised project design and analyses as well as edited manuscript. All the authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Under the Australian Code for the Care and use of animals for scientific purposes, institutional ethics approval involving experimentation on invertebrates (excluding cephalopods) is not required; therefore, animal ethics approval was not required for this population genetics study on the invertebrate Penaeus monodon (see https://www.nhmrc.gov.au/about-us/publications/australian-code-care-and-use-animals-scientific-purposes#block-views-block-file-attachments-content-block-1). No permission or licence was required for the collection of wild adult P. monodon samples used in this study so they were obtained from commercial operators as a result of their normal activities.
Consent for publication
Not applicable.
Competing interests
All authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised: Figure 3 has been updated.
Supplementary information
Additional file 1.
SNP data analysis workflow from raw (n = 125,511) to final neutral (n = 10,535) and outlier (n = 89) loci datasets.
Additional file 2.
Genotypic data. Genotypes of 278 individuals of P. monodon at 10,624 adaptive and neutral genome-wide SNPs are included in a standard STRUCTURE format.
Additional file 3.
Mantel correlograms for the relationship between genetic distance (FST) and geographic distance (km) among Penaeus monodon populations (n = 7) using 10,624 SNPs.
Additional file 4.
Genotypic data. Genotypes of 278 individuals of P. monodon at 89 adaptive genome-wide SNPs are included in a standard STRUCTURE format.
Additional file 5.
Genotypic data. Genotypes of 278 individuals of P. monodon at 10,535 neutral genome-wide SNPs are included in a standard STRUCTURE format.
Additional file 6.
Population differentiation estimates for Penaeus monodon populations sampled using neutral and outlier loci. Population pairwise FST (Weir & Cockerham, 1984) estimates computed by using the R package StAMPP v.1.5.1 (Pembleton et al., 2013). Pairwise FST values of 89 outlier loci are shown below the diagonal, and pairwise FST values of 10,535 neutral loci are reported above. All FST values were significant at p ≤ 0.05 following 1000 bootstraps performed across loci to generate confidence intervals. The only non-significant FST value (p > 0.05) is in bold type.
Additional file 7.
Characterization of high-quality BLAST matches of outlier SNP to Penaeus monodon transcriptome contigs.
Additional file 8.
Translation of eight outlier SNPs with transcriptome matches to protein sequences using all three reading frames.
Additional file 9.
Sampling codes and site locations with number of individuals (N).
Additional file 10.
Marine environmental variables (n = 23) for each of the seven geographically discrete sampling locations as determined using location-specific latitude and longitude.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Vu, N.T.T., Zenger, K.R., Guppy, J.L. et al. Fine-scale population structure and evidence for local adaptation in Australian giant black tiger shrimp (Penaeus monodon) using SNP analysis. BMC Genomics 21, 669 (2020). https://doi.org/10.1186/s12864-020-07084-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-020-07084-x