
Abstract
Background
The contribution of mitogen-activated protein kinase (MAPK) cascades to plant growth and development has been widely studied, but this knowledge has not yet been extended to the medicinal plant Salvia miltiorrhiza, which produces a number of pharmacologically active secondary metabolites.
Results
In this study, we performed a genome-wide survey and identified six MAPKKK kinases (MAPKKKKs), 83 MAPKK kinases (MAPKKKs), nine MAPK kinases (MAPKKs) and 18 MAPKs in the S. miltiorrhiza genome. Within each class of genes, a small number of subfamilies were recognized. A transcriptional analysis revealed differences in the genes’ behaviour with respect to both their site of transcription and their inducibility by elicitors and phytohormones. Two genes were identified as strong candidates for playing roles in phytohormone signalling. A gene-to-metabolite network was constructed based on correlation analysis, highlighting the likely involvement of two of the cascades in the synthesis of two key groups of pharmacologically active secondary metabolites: phenolic acids and tanshinones.
Conclusion
The data provide insight into the functional diversification and conservation of MAPK cascades in S. miltiorrhiza.
Similar content being viewed by others
Background
Plants have developed diverse strategies to protect themselves from pathogens and environmental stress, many of which are based on the production of secondary metabolites [1,2,3,4]. Metabolic engineering of natural product pathways is a feasible strategy over the years for enhancement of plant disease resistance [5]. Some of these compounds also have beneficial nutraceutical or pharmacological properties (classic bacteriostatic, antibiotic, antivirulence, anticancer, anti-diabetic, ect.) [6,7,8,9]. It has been estimated that at least 30% of therapeutic compounds in current use have been derived from medicinal plants [10]. The root of S. miltiorrhiza has a long history of use in Chinese herbal medicine as a source of compounds that are effective for curing a range of illnesses [2]. Its major bioactive compounds fall into two groups: hydrophilic phenolic acids and lipophilic tanshinones [9]. The former are synthesized in planta through both the phenylpropanoid and a tyrosine-derived pathways [11, 12], while the latter are generated through the cytoplasmic mevalonic acid pathway and the plastidial 2-C-methyl-D-erythritol-4-phosphate pathway [13]. Treating plants with the phytohormones have been shown to promote the accumulation of both phenolic acids [11] and tanshinones [13].
Mitogen-activated protein kinase (MAPK) cascades are a universal characteristic of eukaryotic cells. These cascades involve the activity of four distinct types of kinases: MAP kinases (MAPKs), MAPK kinases (MAPKKs), MAPKK kinases (MAPKKKs) and MAPKKK kinases (MAPKKKKs) [14,15,16]. MAPK cascades are important for plant growth [17], development [18,19,20] and defence against biotic [21, 22] and abiotic stress [23,24,25]. Furthermore, MAPK cascades are very likely to be involved in secondary metabolism, including camalexin [26], indole glucosinolate [26], nicotine [27], anthocyanin [28] and phytoalexin [29]. It has been reported that methyl jasmonate (MeJA), salicylic acid (SA), gibberellic acid (GA) and abscisic acid (ABA) regulate the accumulation of phenolic acids [11] and tanshinones [13] in S. miltiorrhiza. Furthermore, the MAPK cascade regulates the biosynthesis and signalling pathways of SA [22, 29, 30], JA [31,32,33], ABA [25, 31, 34,35,36,37], auxin (AUX) [17, 19] and ethylene (ETH) [17, 26]. The acquisition of an increasing number of whole plant genome sequences has revealed large numbers of genes encoding the component enzymes of MAPK cascades. For example, a total of 75 MAPKKK [38], 8 MAPKK [39], and 17 MAPK [40] genes have been reported in the rice genome, whereas the Arabidopsis thaliana genome contains 10 MAPKKKK, 80 MAPKKK, 10 MAPKK and 20 MAPK genes [15, 16]. It was also reported that 74 MAPKKK, 9 MAPKK, and 19 MAPK genes can be found in maize [41,42,43], whereas 89 putative MAPKKK, 6 MAPKK, and 16 MAPK genes are found in tomato [44, 45]. The present study takes advantage of the availability of a complete S. miltiorrhiza genome sequence [46] to document the species’ MAPK cascade gene content and was undertaken because these genes are likely important for the synthesis of its bioactive secondary metabolites. Their identification may lead to the application of metabolic engineering with a view to improving the productivity of the S. miltiorrhiza plant.
Results
The MAPK cascade enzymes encoded by S. miltiorrhiza
The HMMER-based search of the S. miltiorrhiza genome sequence [46] identified a total of six SmMAPKKKKs, 83 SmMAPKKKs, nine SmMAPKKs and 18 SmMAPKs using 10 MAPKKKKs, 80 MAPKKKs, 10 MAPKKs and 20 MAPK sequences from the A. thaliana genome as queries [15, 16]. The relevant sequences and gene ID are provided in Additional file 1 Table S1, while their key structural and other details are provided in Additional file 1 Table S2. The length of the predicted MAPKs ranged from 353 (SmMAPK7) to 690 (SmMAPK10) residues, that of the MAPKKs from 289 (SmMAPKK5) to 521 (SmMAPKK3) residues, that of the MAPKKKs from 185 (SmMAPKKK49) to 1401 (SmMAPKKK3) residues and that of the MAPKKKKs from 456 (SmMAPKKKK6) to 837 (SmMAPKKKK5) residues; the molecular weights in the full set of polypeptides ranged from 32.4 kDa (SmMAPKK5) to 155.3 kDa (SmMAPKKK3), and their pI ranged from 4.49 (SmMAPKKK38) to 9.77 (SmMAPKKK14).
The phylogeny and exon-intron structure of the MAPK cascade genes
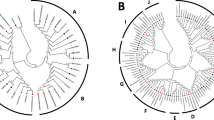
The alignment of S. miltiorrhiza sequences with those of A. thaliana revealed that the SmMAPKs fell into four subfamilies (A through D), the SmMAPKKs into five subfamilies (A through E), the SmMAPKKKs into three subfamilies (MEKK, ZIK and RAF) and the SmMAPKKKKs into two subfamilies (GCK-III and -VI) (Fig. 1). The RAFs constituted the largest single subfamily (38 members), followed by the MEKKs (36 members); only a single SmMAPKK was present in each of subfamilies B, D and E, as was the case for subfamily D of the SmMAPKs. The variation in the exon-intron structure of the S. miltiorrhiza genes is illustrated in Fig. 2. The number of introns present among the SmMAPKs varied from one (SmMAPK9, SmMAPK10) to 11 (SmMAPK6, SmMAPK11, SmMAPK14, SmMAPK18); among the SmMAPKKs from zero (SmMAPKK4, SmMAPKK5, SmMAPKK7 and SmMAPKK9) to eight (SmMAPKK3); among the SmMAPKKKs from zero (SmMAPKKK7, SmMAPKKK8, SmMAPKKK13, SmMAPKKK19, SmMAPKKK21, SmMAPKKK23, SmMAPKKK24, SmMAPKKK25, SmMAPKKK34, SmMAPKKK38, SmMAPKKK39 and SmMAPKKK40) to 19 (SmMAPKKK3) and among the SmMAPKKKKs from zero (SmMAPKKKK6) to 23 (SmMAPKKKK5). The SmMAPKs in subfamily A all harboured six exons and showed similar gene lengths to one another, while the subfamily C members all harboured two exons.

The phylogeny of the SmMAPK gene family. The dendrograms were constructed using the neighbour-joining method applied to full-length A. thaliana and S. miltiorrhiza MAPK sequences. Bootstrap (500 replicates) values appear at each branch. a MAPK sequences, b MAPKK sequences, c MAPKKKK sequences, d MAPKKK sequences

The intron/exon structure of the SmMAPK gene family members. a MAPK sequences, b MAPKK sequences, c MAPKKKK sequences, and d MAPKKK sequences. Exons are shown as yellow boxes and introns with a simple line. Untranslated regions are indicated by thick blue lines. 0, 1, and 2 represent the intron phase. Gene models are drawn to scale
Motif content of SmMAPK enzymes
All of the predicted gene products included various characteristics associated with MAPK cascade enzymes. The SmMAPKs shared the conserved TxY motif (Fig. 3a) contained within the activation loop lying between subdomains VII and VIII as well as the (LH)DxxDE(P) x CD domain (Fig. 3b), which acts as the MAPKK docking site. The TxY motif in the members of subfamilies A, B and C was represented by TEY, and in subfamily D, it was represented by TDY. Each of the SmMAPKKs harboured a D(L/I/V) K motif along with the consensus sequence S/T-× 5-S/T (Fig. 3c). The latter motif was conserved across all members of subfamilies A through D, but the S/T site was altered to G/A in the subfamily E member SmMAPKK8. Among the MAPKKKs, the MEKKs all retained the conserved signature sequence G(T/S)P-x-(W/Y/F) MAPEV, the RAFs retained the GT-x-x-(W/Y) MAPE sequence, and the ZIKs retained the GTPEFMAPE(L/V) Y sequence (Fig. 3d-f). The two motifs shared by the SmMAPKKKKs were H-R/H-D-L/I/V-K-x-x-N/S (subdomain VIb) and G-T/S-x-x-W/Y/F-M/L/−A/S/P-P-E (subdomain VII) (Fig. 3g).

The domain content of the SmMAPK gene family products. a TxY, b CD domain, c D(L/I/V)K S/T-× 5-S/T, d G(T/S)P-x-(W/Y/F)MAPEV, e GT-× 2-(W/Y)MAPE, f GTPEFMAPE(L/V)Y, and g HR/HDL/I/VK-× 2-N/S GT/S-× 2-WMAPE
A MEME-based analysis of the full set of sequences confirmed the identity of each group. Thus, nine out of 16 motifs were common to and conserved in all of the SmMAPK group A proteins, 10 out of 12 in the SmMAPKK group C proteins, 11 out of 20 in the SmMAPKKK ZIKs and 9 out of 13 in all of the GCK-IV SmMAPKKKKs (Fig. 4 and Additional file 2 Figure S1). Seven motifs were conserved across all of the S. miltiorrhiza MAPKKKK proteins, including motifs #1 (G-T/S-x-x-W/Y/F-M/L/−A/S/P-P-E) and #3 (H-R/H-D-L/I/V-K-x-x-N/S). All of the GCK-IV MAPKKKK proteins except for SmMAPKKKK4 retained N-terminal motif #5, and all except for SmMAPKKKK6 retained C-terminal motif #11 (Fig. 4d and Additional file 2 Figure S1). Seven motifs (#1 through #5, #7 and #8) were reasonably well conserved among the SmMAPKs; motifs #9, #12 and #14 were only found in subfamily D members, and motif #15 was only found in subfamily C members (Fig. 4a). There were three conserved motifs (#1, #2 and #6) in the SmMAPKKs, but motif #9 was a subfamily C-specific motif (Fig. 4a). Motif #5 was reasonably well conserved among the SmMAPKKKs (Fig. 4c). There were seven conserved motifs (#1 through #5, #7 and #13) in the SmMAPKKKKs, but motifs #6 and #10 were only found in subfamily GCK-IV (Fig. 4d). The sequences 1500 nt upstream of the S. miltiorrhiza MAPK cascade genes harboured an array of cis acting elements (Additional file 1 Table S3).

The motif content of the SmMAPK gene family products. a-d Putative motifs in a) MAPK, b MAPKK, c MAPKKKK, and d MAPKKK sequences, as predicted by MEME software
Coexpression of genes responsible for the synthesis of phenolic acids and tanshinones
The co-expression analysis directed toward genes (the information on enzymes and transcription factors is listed in Additional file 1 Table S4 and Table S5) encoding key phenolic acid pathway enzymes (SmC4H1, SmCYP98A14, SmHPPR1, SmPAL1, SmRAS1 and SmTAT1), transcription factors (SmAREB1, SmbHLH148, SmbHLH37, SmbHLH51, SmERF115, SmERF1L1, SmMYB111, SmMYB36, SmMYB39, SmMYC2a, SmMYC2b, SmPAP1 and SmTTG1) and members of the SmMAPK family revealed two major clusters: one of these grouped SmAREB1, SmERF115, SmMYB39, SmMYC2b, SmPAL1, SmPAP1 and SmTTG1 with SmMAPK2 and SmMAPK5 through SmMAPK10, while the other comprised SmbHLH148, SmbHLH51, SmbLH37, SmC4H1, SmCYP98A14 SmERF1L1, SmHPPR1, SmMYB111, SmMYB36, SmMYC2a, SmRAS1 and SmTAT1 together with SmMAPK3, SmMAPK4, SmMAPK13 and SmMAPK15 through SmMAPK18 (Fig. 5a). The former group formed two subclusters, one comprising SmERF115, SmMYB39 and SmPAP1 along with the SmMAPK members SmMAPK1, SmMAPK2, SmMAPK6 through SmMAPK9, SmMAPK11 and SmMAPK12; the other linked SmAREB1, SmMYC2b, SmPAL1 and SmTTG1 with SmMAPK5 and SmMAPK10 (Fig. 5a). The second major cluster formed four subclusters; in one of these subclusters, SmMAPK3 was grouped with SmbHLH51, SmC4H1, SmCYP98A14, SmERF11, SmHPPR1, SmMYB111, SmMYC2a, SmRAS1 and SmTAT1, while the other important subcluster grouped SmbHLH37 and SmbHLH148 with SmMAPK4, SmMAPK13 and SmMAPK15 through SmMAPK17 (Fig. 5a). The similarity between the transcriptional behaviour of SmbHLH51 and SmMAPK3 implies that, given that SmbHLH51 has been identified as a positive transcriptional regulator of phenolic acid synthesis [47], SmMAPK3 very likely functions within the phenolic acid synthesis pathway (Fig. 5a).

Heat maps illustrating patterns of gene coexpression. Genes encoding (a) key phenolic acid pathway enzymes, transcription factors and members of the SmMAPK family, b enzymes involved in the synthesis of tanshinones, transcription factors and members of the SmMAPK family. Transcript abundance was estimated in the roots, leaves and flowers of S. miltiorrhiza plants, some of which were exposed to salicylic acid, methyl jasmonate or yeast extract. Pearson correlation coefficient (PCC) values were calculated for these genes. Blue: low abundance, red: high abundance
The coexpression analysis directed toward genes encoding enzymes involved in the synthesis of tanshinones (SmCPS1, SmCPS2, SmCYP76AH1, SmCYP76AH3, SmCYP76AK1, SmDXR, SmGGPPS1, SmHMGR1, SmHMGR2, SmIPI1 and SmKSL1), transcription factors (SmAREB1, SmbHLH10, SmbHLH148, SmERF115, SmERF1L1, SmERF6, SmMYB36, SmMYB9, SmMYC2a, SmMYC2b and SmWRKY1) and members of the SmMAPK family revealed a transcriptional relationship between SmMAPK1, SmMAPK2, SmMAPK5 through SmMAPK12, SmAREB1, SmCPS1, SmCPS2, SmCYP76AH1, SmCYP76AH3, SmCYP76AK1, SmDXR, SmERF5, SmGGPPS1, SmHMGR2, SmKSL1, SmMYC2 and SmWRKY1 (Fig. 5b). Further linkage was noted between SmMAPK3, SmMAPK4, and SmMAPK13 through SmMAPK18 and SmbHLH10, SmbHLH148, SmERF1L1, SmERF6, SmHMGR1, SmIPI1, SmMYB36, SmMYB9 and SmMYC2a (Fig. 5b). The former cluster resolved into three major subclusters. One of these comprised the genes encoding enzymes acting throughout the mevalonic acid and 2-C-methyl-D-erythritol-4-phosphate pathways (SmCPS1, SmCYP76AH1, SmCYP76AH3, SmCYP76AK1, SmDXR, SmGGPPS1, SmHMGR2 and SmKSL1) along with SmERF115 and the SmMAPK members SmMAPK1, SmMAPK2, SmMAPK6 through SmMAPK9, SmMAPK11 and SmMAPK12). The second cluster grouped genes encoding two upstream enzymes (SmHMGR1 and SmIPI1) with those encoding seven transcription factors (SmbHLH10, SmbHLH148, SmERF1L1, SmERF6, SmMYB36, SmMYB9 and SmMYC2a). Similar patterns of transcription were shown by the gene pairs SmAREB1/SmMAPK5, SmWRKY1/SmMAPK10, SmERF115/SmMAPK2, SmHMGR1/SmMAPK16, SmbHLH148/SmMAPK13, SmbHLH10/SmMAPK14 and SmMYB36/SmMAPK18 (Fig. 5b), implying the involvement of some SmMAPK products in the synthesis of tanshinones. The transcriptional behaviour of SmIPI1, SmMYB9 and SmMAPK3 was also quite similar.
As shown in Fig. 5, the expression of SmMAPK14 (Group D) in different tissues/treatments was not significantly different. In addition, some gene expression preferences in different tissues/treatments could be observed from RNA-Seq data. For example, the expression of SmMAPK4 (Group B) in different tissues (roots, flowers and leaves) and treatments (MeJA and YE) was more obvious than that under treatment with SA. Several group D members (SmMAPK13, SmMAPK15 and SmMAPK17) showed higher gene expression levels in roots than in the treatments and other tissues, and SmMAPK18 presented higher gene expression levels in flowers and leaves and under SA treatment. Interestingly, gene members in group A (SmMAPK1, SmMAPK2 and SmMAPK3) presented higher expression levels than those in the other groups (Group B, Group C, and Group D). There are also mounting concerns that the expression of SmMAPK3 in the roots and under treatment with MeJA, YE and SA is biased. In contrast, the expression of SmMAPK1 in roots was lower than that under the treatments and in other tissues, and the expression of SmMAPK2 was reduced after short-term treatments. This means that gene members in group A show complex responses to hormone treatments and complex regulatory mechanisms of phenolic acid and tanshinone synthesis under hormone treatments.
Coexpression analysis of SmMAPKs and likely MAPK cascades acting in S. miltiorrhiza
The results of the coexpression analysis of the SmMAPKs family are shown in Fig. 6a, and the interaction network of the MAPK cascades is presented in Additional file 2 Figure S2. Based on the situation in A. thaliana, the expectation was that there would also be two MAPK cascades in S. miltiorrhiza, one related to AtMPK6 and the other to AtMPK3. However, the analysis implied that there were three, related to SmMAPK1 (84.8% similar to AtMPK6), SmMAPK2 (likely homologue of AtMPK6) and SmMAPK3 (likely homologue of AtMPK3). The interaction network shown in Fig. 6b involved two cascades: the participants in the first were SmMAPK1, SmMAPKK3, four SmMAPKKKs (SmMAPKKK29, SmMAPKKK59, SmMAPKKK63, SmMAPKKK82) and four SmMAPKKKKs (SmMAPKKKK1, SmMAPKKKK3 through SmMAPKKKK5), and those in the second were SmMAPK3, SmMAPKK2, eight SmMAPKKKs (SmMAPKKK3, SmMAPKKK32, SmMAPKKK41, SmMAPKKK51, SmMAPKKK57, SmMAPKKK59, SmMAPKKK64 and SmMAPKKK83) and three SmMAPKKKKs (SmMAPKKKK3 through SmMAPKKKK5) (Fig. 6c). SmMAPK1 might be activated by SmMAPKK3, which is in turn probably activated by SmMAPKKK29, SmMAPKKK59, SmMAPKKK63 and SmMAPKKK82; these MAPKKKs could be activated by MAPKKK kinases (SmMAPKKKK1 and SmMAPKKKK3 through SmMAPKKKK5). On the other hand, SmMAPKK2 might be activated by the MAPKK kinases SmMAPKKK3, SmMAPKKK32, SmMAPKKK41, SmMAPKKK51, SmMAPKKK57, SmMAPKKK59, SmMAPKKK64 and SmMAPKKK83, which are in turn probably activated by the MAPKKK kinases SmMAPKKKK1 and SmMAPKKKK3 through SmMAPKKKK5. These proteins phosphorylate SmMAPKK2, which in turn phosphorylates SmMAPK3 (Fig. 6c).

Probable MAPK cascades in S. miltiorrhiza. a Heat maps illustrating patterns of gene coexpression generated by Multi Experiment Viewer. Blue: low abundance, red: high abundance. b Network built on the basis of the correlation among SmMAPKKKKs, SmMAPKKKs, SmMAPKKs and SmMAPKs. Pearson correlation coefficient (PCC) values were calculated for each pair of genes. MAPK cascade reaction map constructed using Cytoscape 3.6.1.0 software. c MAPK cascade pattern diagram
To validate the results of the coexpression analysis, we used quantitative RT-PCR. A total of 30 genes were tested, including MAPK cascade genes, enzymes and transcription factors (TFs). The correlation coefficient of the R value between the Ct value of the qRT-PCR results and the log2 RPKM values from the RNA-seq analysis was calculated for each gene via Pearson correlation. The results are presented in Additional file 1 Figure S3. It was clear that the expression of SmMAPK3 according to qRT-PCR was similar to that according to RNA-Seq (Fig. 7). Furthermore, TFs (SmERF6/SmERF115) and enzymes (SmIPI1/SmHMGR2/SmDXR/SmCYP98A14) showed the same pattern (Fig. 7). In addition, Fig. 7 shows that the pattern differed between the qRT-PCR and RNA-seq data for some genes, such as SmMAPK3 in roots, SmMYB36 in roots and leaves and SmMAPK6 under YE treatment. The minor difference between the qRT-PCR and RNA-seq might be caused by two experimental systems. It was normal also because the site where we harvested the plant material differed from the site where the materials used to generat the RNA-seq data were collected. This is an inevitable error because we could not obtain the same samples used in the other analyses.

qRT-PCR analyses of coexpressed genes. a Heat maps of coexpressed genes based on qRT-PCR analyses. b Heat maps of coexpressed genes based on RNA-Seq analyses
SmMAPK3 directly interacts with SmMYBs and SmAREB1
For the Y2H assay, SmMAPK3 was fused to pGBKT7, and SmMYC2a, SmMYC2b, SmMYB36, SmMYB39, SmMYB111, SmMYB9b, SmPAP1, SmTTG1, SmWRKY1, SmAREB1, SmERF6, SmHLH51, SmHLH10, SmHLH148, SmHLH37 and SmERF1L1 were ligated to pGADT7 to generate pGBKT7-SmMAPK3 and pGADT7-TF. The Y2H Gold strain yeast cells transformed with BD-SmMAPK3 and AD-SmMYC2a/SmMYC2b/SmMYB9b/SmTTG1/SmWRKY1/SmERF6/SmHLH51/SmHLH10/SmHLH148/SmHLH37/SmERF1L1 could not grow on SD-LWHA with AbA and X-ɑ-Gal, while the Y2H Gold strain yeast cells cotransformed with BD-SmMAPK3 and AD-SmMYB36/SmMYB39/SmMYB111/SmPAP1/SmAREB1 could grow on SD-LWHA with AbA and expressed Mel1 and turned blue in the presence of the X-α -Gal substrate (Fig. 8).

SmMAPK3 physically interacts with SmMYBs and SmAREB1. Y2H assay to detect the interactions of SmMAPK3 with SmAREB1, SmbHLH10, SmbHLH37, SmbHLH51, SmbHLH148, SmERF1L1, SmERF6, SmMYB9b, SmMYB36, SmMYB39, SmMYB111, SmMYC2a, SmMYC2b, SmPAP1, SmTTG1 and SmWRKY1. Transformed yeast was grown on selective medium lacking adenine, histidine, leucine, and tryptophan (SD-LWHA) with AbA and x-a-gal to test protein interactions
Discussion
A number of studies have focused on systematically identifying and characterizing the proteins participating in plant MAPK cascades [48,49,50,51], which affect various aspects of both growth and development [18, 52, 53], stress tolerance [23, 24, 34, 54] and the synthesis of secondary metabolites [17, 26, 33]. The present research has extended the scope of these studies to the medicinal species S. miltiorrhiza, made possible by the recent acquisition of its genomic sequence [46]. Our signal outcome was the identification of six SmMAPKKKKs, 83 SmMAPKKKs, nine SmMAPKKs, and 18 SmMAPK genes containing conserved domains through genome-wide analysis. At the same time, cascades involving the participation of SmMAPK3 and SmMAPK1 were identified, which were most likely to be involved in phenolic acid and tanshinone biosynthesis according to an enzyme and TF gene-to-MAPK cascade gene correlation analysis.
Through the exploitation of RNA-seq-based data, it was possible to correlate the transcriptional profiles of genes encoding both key enzymes and transcription factors with members of the SmMAPK gene family. For example, the profiles of the two enzyme-encoding genes SmHMGR1 and SmIPI1 resembled those of seven transcription factor-encoding genes (SmbHLH10, SmbHLH148, SmERF1L1, SmERF6, SmMYB36, SmMYB9 and SmMYC2a) (Fig. 5b). Note that SmIPI1 has been identified as a key enzyme in the terpenoid metabolic pathway [55]; the correlation of the transcription of SmMAPK3 with that of SmIPI1 suggested that the product of the former gene may be involved in the regulation of tanshinone synthesis. The product of SmERF6 modulates the synthesis of tanshinones through its binding to a GCC box present in the promoters of both SmCPS1 and SmKSL1 [56], while SmMYB36 interacts with many MYB-related core elements (MBSI, MBSII, AAAAAAC(C/G) GTTA, CAACTG and AAAAGTTAGTTA) present in the promoters of various genes encoding enzymes involved in the synthesis of secondary metabolites [57]. Correlations were also identified for a set of eight enzyme-encoding genes (SmCPS1, SmCYP76AH1, SmCYP76AH3, SmCYP76AK1, SmDXR, SmGGPPS1, SmHMGR2 and SmKSL1) along with three genes encoding transcription factors (SmAREB1, SmERF115 and SmWRKY1). SmWRKY1 has been shown to participate in the regulation of tanshinone synthesis through its interaction with SmDXR [58]. Another set of correlated genes included six genes encoding transcription factors (SmAREB1, SmERF115, SmMYB3, SmMYC2b, SmPAP1 and SmTTG1) and one gene encoding an enzyme (SmPAL1) (Fig. 5a). SmAREB1 has been demonstrated to promote the flux of metabolites through the phenolic acid-branched pathway via its phosphorylation of SmSnRK2.6 [59]. The overexpression of SmMYC2 promotes the production of Sal B [60]. AtMPK6, which is phosphorylated by AtMKK3 (MAPK kinase 3), phosphorylates a basic helix-loop-helix transcription factor, AtMYC2. Furthermore, AtMYC2 binds to the AtMPK6 promoter and regulates its expression in a feedback regulatory mechanism in blue light signalling [61]. Therefore, SmMAPK cascades probably phosphorylate SmMYC2 or other TFs, and the TFs then regulate the expression of enzyme genes. SmMAPK2, SmMAPK3, SmMAPK5, SmMAPK8, SmMAPK10, SmMAPK13 and SmMAPK18 shared a similar transcriptional profile with a number of genes encoding either enzymes or transcription factors associated with the synthesis of phenolic acids, as did SmMAPK2, SmMAPK3, SmMAPK5, SmMAPK10, SmMAPK13, SmMAPK14, SmMAPK16 and SmMAPK18 with genes encoding either enzymes or transcription factors associated with the synthesis of tanshinones (Fig. 5) Nine of the 18 SmMAPK proteins (SmMAPK2, SmMAPK3, SmMAPK5, SmMAPK8, SmMAPK10, SmMAPK13, SmMAPK14, SmMAPK16 and SmMAPK18) are potentially involved in the synthesis of key secondary metabolites; six of these proteins (SmMAPK2, SmMAPK3, SmMAPK5, SmMAPK10, SmMAPK13 and SmMAPK18) potentially participate in the synthesis of both phenolic acids and tanshinones, while SmMAPK8 is involved in the synthesis of only phenolic acids, and SmMAPK14 and SmMAPK16 are involved in the synthesis of nonphenolic acids but not phenolic acids. It is proposed that SmMAPK2, SmMAPK3, SmMAPK5, SmMAPK8 and SmMAPK10 are positive regulators, while SmMAPK18 is a negative regulator of phenolic acid synthesis; SmMAPK3, SmMAPK5, SmMAPK10, SmMAPK13, SmMAPK14, SmMAPK16 and SmMAPK18 also act to promote tanshinone synthesis.
Elicitors such as yeast elicitors, metal ions (Ag+, Cu2+, Zn2+, Co2+), plant growth regulators (SA, ABA, ETH, MeJA, IAA, NAA, GA, 6-BA, TDZ), and other treatments (polyamines, ultraviolet-B radiation, H2O2), induce the biosynthesis and accumulation of secondary metabolites (tanshinone and phenolic acid biosynthesis) in S. miltiorrhiza [11, 13], especially plant hormones (MeJA, SA, ETH, ABA, GA) [62,63,64]. MAPK cascades have also been implicated in ABA, ETH, JA and SA signalling [17, 25, 29, 33, 36, 37]. ABA, ETH, JA and SA are important plant hormones, and their crosstalk is crucial for secondary metabolite biosynthesis during defence against pathogens and insects [65, 66]. The present analysis suggested that in S. miltiorrhiza, members of group A (SmMAPK1, SmMAPK2 and SmMAPK3) show a higher expression level under treatments with MeJA, YE and SA than the other groups (Group B, Group C, and Group D) (Fig. 5), and SmMAPK1 and SmMAPK3 are both probably important regulators of secondary metabolite synthesis (Fig. 6c). SmMAPK3 can physically interact with SmMYB36/SmMYB39/SmMYB111/SmPAP1/SmAREB1 (Fig. 8), which have been reported to regulate the synthesis and accumulation of secondary metabolites [57, 59, 67,68,69]. In A. thaliana, MAPKK9 promotes ethylene and camalexin biosynthesis [70], and both AtMPK3 and AtMPK6, which are highly involved in the plant response to biotic [71,72,73,74,75] and abiotic stress [21, 54, 76] and the regulation of ETH [17, 26, 33], SA [29, 75] and JA [33, 77] production, are activated by the upstream regulatory MAPK kinases AtMKK4 and AtMKK5 [78, 79], which are in turn regulated by the upstream MAPKK kinase AtMEKK1 [80]. Such interspecific similarity in MAPK function and interspecific differences in the architecture of MAPK cascades explain the conservation and variability of gene evolution.
To further identify and characterize the functions of candidate genes (such as SmMAPK3), we plan to overexpress and conducted RNA interference knockdown of SmMAPK3 to obtain an initial understanding of its function in phenolic acid and tanshinone synthesis. Thereafter, we will validate the proteins interacting with SmMAPK3 (either screened in Y2H assays or reported in the literature). Then, we will verify the function of the interacting proteins and determine their upstream and downstream relationships. Ultimately, we will be able to improve the quality of S. miltiorrhiza through genetic modification.
Conclusions
The present study has provided an exhaustive catalogue of the genes encoding MAPKs, MAPKKs, MAPKKKs and MAPKKKKs in S. miltiorrhiza: 18 genes were identified as putatively encoding MAPKs, nine MAPKKs, 83 MAPKKKs and six MAPKKKKs. The set of SmMAPKKKKs formed two subfamilies (GCK-III, GCK-VI), the SmMAPKKKs three subfamilies (MEKK, ZIK and RAF), the SmMAPKKs five subfamilies (A through E) and the SmMAPKs four subfamilies (groups A through D). The identity of each subfamily was supported by its sequence-based phylogeny, by the exon-intron structure of its member genes and by the content of conserved domains/motifs. Most of these genes were transcriptionally active in the roots, leaves and flowers of the S. miltiorrhiza plant, although there was some evidence of topological specialization of a few of these genes. The results of quantitative RT-PCR verified that the gene coexpression analysis based on the RNA-seq data was accurate. Group A of the SmMAPK genes appeared to be inducible, not only by elicitors such as yeast extract but also by phytohormones such as salicylic acid and jasmonate. They appear to be prominently expressed during the defence response and the synthesis of secondary metabolites. According to the Y2H assay, SmMAPK3 physically binds TFs including SmMYB36, SmMYB39, SmMYB111, SmPAP1 and SmAREB1, probably to regulate the synthesis of secondary metabolites.
Methods
Plant materials
Three different S. miltiorrhiza tissues, the roots, flowers, and leaves, were collected from the botanical garden of S. miltiorrhiza at Northwest A&F University in Yangling, China. S. miltiorrhiza hairy roots (0.3 g FW) resulting from the infection of sterile plantlets with Agrobacterium rhizogenes (ATCC15834) (WEIDI, Shanghai, China) were cultured in a 100 ml shake flask containing 50 ml of liquid 6,7-V medium on an orbital shaker.
Gene discovery and bioinformatic analysis
Hidden Markov model profiles were built using HMMER v.3.1 software [81] (www.hmmer.org) with the aim of identifying MAPK cascade family sequences in the S. miltiorrhiza genome. The query sequences from A. thaliana (the sequences and Accession Numbers are in Additional file 1 Table S6) comprised 20 genes encoding MAPKs, 10 MAPKKs, 80 MAPKKKs and 10 MAPKKKKs [15, 16]. The applied E value threshold was 10− 6. After the manual removal of redundant sequences, alignment was performed using the Clustal W program [82]. A phylogenetic analysis was conducted using MEGA v7.0 software [83] (www.megasoftware.net) applying the neighbour-joining method [84] and a 500 replication bootstrap test. The molecular weight and pI of each gene product were predicted using the Compute pI/MW tool mounted on the ExPASy server [85] (www.expasy.org). The members of MAPK cascades were also subjected to analysis based on MEME software [86] (meme-suite.org/tools/meme). Gene structures were obtained using Gene Structure Display Server 2.0 (gsds.cbi.pku.edu.cn/). Conserved domains were identified using DNAMAN software (https://www.lynnon.com/). Sequences upstream of the transcription start site (ATG) of the SMMAPKs were derived from the S. miltiorrhiza genome sequence. The cis-acting element content of these promoter-containing sequences was deduced using the PLACE database (www.dna.affrc.go.jp/PLACE/signalscan.html).
Coexpression analyses
RNA-seq reads were derived from mRNA extracted from the leaves, flowers and roots of plants subjected to three treatments (salicylic acid, methyl jasmonate and yeast extract). The reads were recovered from the Sequence Read Archive (SRA) (www.ncbi.nlm.nih.gov/sra) under accession numbers SRR1043998, SRR1045051, SRR1020591, SRX1423774, SRX2992229, SRX2992230, SRX2992231, SRX2992232 and SRX2992233. RPKMs, calculated using the BMKCloud tool (www.biocloud.net), were used to derive the Pearson correlation coefficient for each pair of transcripts using the bivariate correlation analysis tool implemented in Excel2010; correlations > 0.5 were considered significant.
Coexpression/coregulation cluster analysis was performed for samples from 12 different tissues or time point by MeV (Version 4.9) [87]. The normalized expression values of the genes were calculated by dividing their expression levels from different tissues or time points. Hierarchical clustering (HCL) was performed using MeV with default settings. The MAPK cascade reaction map was constructed with Cytoscape 3.6.1.0 software (https://cytoscape.org/).
RNA extraction and gene expression analysis
For the quantitative real-time PCR (qRT-PCR) analysis of MAPK cascade genes and their coexpressed genes in S. miltiorrhiza, total RNA was extracted from the leaves, flowers, and roots of plants and Danshen hairy roots treated for different times with salicylic acid, methyl jasmonate and yeast extract [88]. Total RNA was isolated by using the RNAprep Pure Plant Kit (TIANGEN, Beijing, China) according to the manufacturer’s instructions. cDNA was prepared from total RNA by using the PrimeScript RT reagent kit (TaKaRa, Dalian, Chain). For every sample, qRT-PCR was performed on a real-time PCR system (Bio-RAD CFX96, CA, USA) with the TB Green® Premix Ex TaqTM II Kit (TaKaRa, China). Gene-specific primers (Additional file 1 Table S7) were designed with Primer Premier v5.0 software to detect the expression of relevant genes. The expression levels of target genes were normalized to those of β-actin and ubiquitin [59].
Yeast two-hybrid (Y2H) assays
The coding sequence of the SmMAPK3 gene was cloned into the pGBKT7 vector, and those of SmMYC2a, SmMYC2b, SmMYB36, SmMYB39, SmMYB111, SmMYB9b, SmPAP1, SmTTG1, SmWRKY1, SmAREB1, SmERF6, SmHLH51, SmHLH10, SmHLH148, SmHLH37 and SmERF1L1 were cloned into pGADT7. The plasmids were transformed into Y2H Gold yeast cells and grown on SD-dropout medium lacking leucine and tryptophan (SD-LW) medium. Furthermore, yeast cells were screened on SD-selection medium lacking adenine, histidine, leucine, and tryptophan (SD-LWHA) with aureobasidin A (AbA) and a-galactosidase (X-ɑ-Gal). Interactions were observed after a 3 d of incubation at 30 °C.
Statistical analysis
All statistical calculations were performed using routines implemented in SPSS v18.0 software; the chosen significance thresholds were P < 0.05 and < 0.01.
Availability of data and materials
The DNA sequencing data and the Protein sequences are available in the National Data Center of Traditional Chinese Medicine of China database with the link of ftp://danshen.ndctcm.org:10402/, under the gene ID provided in Additional file 1 Table S1. The query sequences from A. thaliana are available in the National Center for Biotechnology Information (NCBI) database with the link of https://www.ncbi.nlm.nih.gov/, under the accession number provided in Additional file 1 Table S6. And the RNA sequencing data are available in the Sequence Read Archive (SRA) database with the link of www.ncbi.nlm.nih.gov/sra, under the accession number SRR1043998, SRR1045051, SRR1020591, SRX1423774, SRX2992229, SRX2992230, SRX2992231, SRX2992232 and SRX2992233, respectively. All of the datasets supporting the results of this article are included within the article and its Additional files.
Abbreviations
- ABA:
-
Abscisic acid
- C4H:
-
Cinnamate 4-hydroxylase
- HPPR:
-
4-Hydroxyphenylpyruvate reductase
- JA:
-
Jasmonic acid
- MAPK cascades:
-
Mitogen-activated protein kinase cascades
- MAPK:
-
Mitogen-activated protein kinase
- MAPKK:
-
MAPK kinase
- MAPKKK:
-
MAPK kinase kinase
- MAPKKKK:
-
MAPK kinase kinase kinase kinase
- MeJA:
-
Methyl jasmonate
- MEME:
-
Multiple expectation maximization for motif elicitation
- NCBI:
-
National Center for Biotechnology Information
- PAL:
-
Phenylalanine ammonia-lyase
- RAS:
-
Rosmarinic acid synthase
- SA:
-
Salicylic acid
- TAIR:
-
The Arabidopsis information resource
- TAT:
-
Tyrosine aminotransferase
- 4CL:
-
4-Coumaric acid CoA-ligase
References
Hidalgo W, Chandran JN, Menezes RC, Otálvaro F, Schneider B. Phenylphenalenones protect banana plants from infection byMycosphaerella fijiensis and are deactivated by metabolic conversion. Plant Cell Environ. 2016;39(3):492–513..
Wu Y, Ni Z, Shi Q, Dong M, Kiyota H, Gu Y, Cong AB. Constituents from Salvia Species and Their Biological Activities. Chem Rev. 2012;112(11):5967–6026.
Petersen M, Abdullah Y, Benner J, Eberle D, Gehlen K, Hücherig S, Janiak V, Kim KH, Sander M, Weitzel C, et al. Evolution of rosmarinic acid biosynthesis. Phytochemistry. 2009;70(15–16):1663–79.
La Camera S, Gouzerh G, Dhondt S, Hoffmann L, Fritig B, Legrand M, Heitz T. Metabolic reprogramming in plant innate immunity: the contributions of phenylpropanoid and oxylipin pathways. Immunol Rev. 2004;198:267–84.
Dixon RA. Natural products and plant disease resistance. Nature. 2001;411(6839):843–7.
Salam AM, Quave CL. Opportunities for plant natural products in infection control. Curr Opin Microbiol. 2018;45:189–94.
Pojero F, Poma P, Spanò V, Montalbano A, Barraja P, Notarbartolo M. Targeting multiple myeloma with natural polyphenols. Eur J Med Chem. 2019;180:465–85.
Ngo YL, Lau CH, Chua LS. Review on rosmarinic acid extraction, fractionation and its anti-diabetic potential. Food Chem Toxicol. 2018;121:687–700.
Wang X, Morris-Natschke SL, Lee KH. New developments in the chemistry and biology of the bioactive constituents of Tanshen. Med Res Rev. 2007;27(1):133–48.
Thiericke R. Drug discovery from nature: automated high-quality sample preparation. J Autom Methods Manag Chem. 2000;22(5):149–57.
Ma P, Liu J, Zhang C, Liang Z. Regulation of water-soluble phenolic acid biosynthesis in Salvia miltiorrhiza Bunge. Appl Biochem Biotech. 2013;170(6):1253–62.
Petersen M, Simmonds MSJ. Rosmarinic acid. Phytochemistry. 2003;62(2):513–7.
Ma P, Liu J, Osbourn A, Dong J, Liang Z. Regulation and metabolic engineering of tanshinone biosynthesis. RSC Adv. 2015;5:18137–44.
Zhang M, Chiang Y, Toruno TY, Lee D, Ma M, Liang X, Lal NK, Lemos M, Lu Y, Ma S, et al. The MAP4 kinase SIK1 ensures robust extracellular ROS burst and antibacterial immunity in plants. Cell Host Microbe. 2018;24:379–91.
Champion A, Picaud A, Henry Y. Reassessing the MAP3K and MAP4K relationships. Trends Plant Sci. 2004;9(3):123.
Ichimura K. Mitogenactivated protein kinase cascades in plant: a new nomenclature. Trends Plant Sci. 2002;7(7):301–8.
Zhang M, Su J, Zhang Y, Xu J, Zhang S. Conveying endogenous and exogenous signals: MAPK cascades in plant growth and defense. Curr Opin Plant Biol. 2018;45:1–10.
Xue X, Bian C, Guo X, Di R, Dong J. The MAPK substrate MASS proteins regulate stomatal development in Arabidopsis. PLoS Genet. 2020;16(4):e1008706.
Chen L, Yang D, Zhang Y, Wu L, Zhang Y, Ye L, Pan C, He Y, Huang L, Ruan Y, et al. Evidence for a specific and critical role of mitogen-activated protein kinase 20 in uni-to-binucleate transition of microgametogenesis in tomato. New Phytol. 2018;219(1):176–94.
Sun T, Nitta Y, Zhang Q, Wu D, Tian H, Lee JS, Zhang Y. Antagonistic interactions between two MAP kinase cascades in plant development and immune signaling. EMBO Rep. 2018;19(7):e45324.
Bigeard J, Hirt H. Nuclear signaling of plant MAPKs. Front Plant Sci. 2018;9:469.
Wang C, He X, Li Y, Wang L, Guo X, Guo X. The cotton MAPK kinase GhMPK20 negatively regulates resistance to Fusarium oxysporum by mediating the MKK4-MPK20-WRKY40 cascade. Mol Plant Pathol. 2018;19(7):1624–38.
Ding H, He J, Wu Y, Wu X, Ge C, Wang Y, Zhong S, Peiter E, Liang J, Xu W. The tomato mitogen-activated protein kinase SlMPK1 is as a negative regulator of the high-temperature stress response. Plant Physiol. 2018;177(2):633–51.
Zhao C, Wang P, Si T, Hsu C, Wang L, Zayed O, Yu Z, Zhu Y, Dong J, Tao WA, et al. MAP kinase cascades regulate the cold response by modulating ICE1 protein stability. Dev Cell. 2017;43(5):618–29.
de Zelicourt A, Colcombet J, Hirt H. The role of MAPK modules and ABA during abiotic stress signaling. Trends Plant Sci. 2016;21(8):677–85.
Thulasi Devendrakumar K, Li X, Zhang Y. MAP kinase signalling: interplays between plant PAMP- and effector-triggered immunity. Cell Mol Life Sci. 2018;75(16):2981–9.
De Boer K, Tilleman S, Pauwels L, Vanden Bossche R, De Sutter V, Vanderhaeghen R, Hilson P, Hamill JD, Goossens A. APETALA2/ETHYLENE Response factor and basic helix-loop-helix tobacco transcription factors cooperatively mediate jasmonate-elicited nicotine biosynthesis. Plant J. 2011;66(6):1053–65.
Lei L, Li Y, Wang Q, Xu J, Chen Y, Yang H, Ren D. Activation of MKK9-MPK3/MPK6 enhances phosphate acquisition in Arabidopsis thaliana. New Phytol. 2014;203(4):1146–60.
Genot B, Lang J, Berriri S, Garmier M, Gilard F, Pateyron S, Haustraete K, Van Der Straeten D, Hirt H, Colcombet J. Constitutively active Arabidopsis MAP kinase 3 triggers defense responses involving salicylic acid and SUMM2 resistance protein. Plant Physiol. 2017;174(2):1238–49.
Xu H, Zhang C, Li Z, Wang Z, Jiang X, Shi Y, Tian S, Braun E, Mei Y, Qiu W, et al. The MAPK kinase kinase GmMEKK1 regulates cell death and defense responses. Plant Physiol. 2018;178(2):907–22.
Mine A, Berens ML, Nobori T, Anver S, Fukumoto K, Winkelmüller TM, Takeda A, Becker D, Tsuda K. Pathogen exploitation of an abscisic acid- and jasmonate-inducible MAPK phosphatase and its interception byArabidopsis immunity. Proc Natl Acad Sci. 2017;114(28):7456–61.
Liu Y, Schiff M, Dinesh-Kumar SP. Involvement of MEK1 MAPKK, NTF6 MAPK, WRKY/MYB transcription factors, COI1 and CTR1 in N -mediated resistance to tobacco mosaic virus. Plant J. 2004;38(5):800–9.
Meng X, Zhang S. MAPK cascades in plant disease resistance signaling. Annu Rev Phytopathol. 2013;51:245–66.
Zhu D, Chang Y, Pei T, Zhang X, Liu L, Li Y, Zhuang J, Yang H, Qin F, Song C, et al. The MAPK-like protein 1 positively regulates maize seedling drought sensitivity by suppressing ABA biosynthesis. Plant J. 2020;102(4):747–60.
Mine A, Berens ML, Nobori T, Anver S, Fukumoto K, Winkelmuller TM, Takeda A, Becker D, Tsuda K. Pathogen exploitation of an abscisic acid- and jasmonate-inducible MAPK phosphatase and its interception by Arabidopsis immunity. Proc Natl Acad Sci U S A. 2017;114(28):7456–61.
Lee Y, Kim YJ, Kim M, Kwak JM. MAPK cascades in guard cell signal transduction. Front Plant Sci. 2016;7:80.
Danquah A, de Zelicourt A, Colcombet J, Hirt H. The role of ABA and MAPK signaling pathways in plant abiotic stress responses. Biotechnol Adv. 2014;32(1):40–52.
Rao KP, Richa T, Kumar K, Raghuram B, Sinha AK. In Silico analysis reveals 75 members of mitogen-activated protein kinase kinase kinase gene family in Rice. DNA Res. 2010;17(3):139–53.
Wankhede DP, Misra M, Singh P, Sinha AK. Rice mitogen activated protein kinase kinase and mitogen activated protein kinase interaction network revealed by in-silico docking and yeast two-hybrid approaches. PLoS One. 2013;8(5):e65011.
Rohila JS, Yang Y. Rice mitogen-activated protein kinase gene family and its role in biotic and abiotic stress response. J Integr Plant Biol. 2007;49(6):751–9.
Kong X, Lv W, Zhang D, Jiang S, Zhang S, Li D. Genome-wide identification and analysis of expression profiles of maize mitogen-activated protein kinase kinase kinase. PLoS One. 2013;8(2):e57714.
Kong X, Pan J, Zhang D, Jiang S, Cai G, Wang L, Li D. Identification of mitogen-activated protein kinase kinase gene family and MKK–MAPK interaction network in maize. Biochem Bioph Res Co. 2013;441(4):964–9.
Sun W, Chen H, Wang J, Sun HW, Yang SK, Sang YL, Lu XB, Xu XH. Expression analysis of genes encoding mitogen-activated protein kinases in maize provides a key link between abiotic stress signaling and plant reproduction. Funct Integr Genomic. 2015;15(1):107–20.
Kong F, Wang J, Cheng L, Liu S, Wu J, Peng Z, Lu G. Genome-wide analysis of the mitogen-activated protein kinase gene family in Solanum lycopersicum. GENE. 2012;499(1):108–20.
Wu J, Wang J, Pan C, Guan X, Wang Y, Liu S, He Y, Chen J, Chen L, Lu G. Genome-wide identification of MAPKK and MAPKKK gene families in tomato and transcriptional profiling analysis during development and stress response. PLoS One. 2014;9(7):e103032.
Xu H, Song J, Luo H, Zhang Y, Li Q, Zhu Y, Xu J, Li Y, Song C, Wang B, et al. Analysis of the genome sequence of the medicinal plant Salvia miltiorrhiza. Mol Plant. 2016;9(6):949–52.
Wu Y, Zhang Y, Li L, Guo X, Wang B, Cao X, Wang Z. AtPAP1 interacts with and activates SmbHLH51, a positive regulator to phenolic acids biosynthesis in Salvia miltiorrhiza. Front Plant Sci. 2018;9:1687.
Jiang M, Chu Z. Comparative analysis of plant MKK gene family reveals novel expansion mechanism of the members and sheds new light on functional conservation. BMC Genomics. 2018;19(1):407.
Kalapos B, Hlavová M, Nádai TV, Galiba G, Bišová K, Dóczi R. Early Evolution of the Mitogen-Activated Protein Kinase Family in the Plant Kingdom. Sci Rep UK. 2019;9(1):4094.
Zhang J, Wang X, Wang Y, Chen Y, Luo J, Li D, Li X. Genome-wide identification and functional characterization of cotton (Gossypium hirsutum) MAPKKK gene family in response to drought stress. BMC Plant Biol. 2020;20(1):217.
Wu P, Wang W, Li Y, Hou X. Divergent evolutionary patterns of the MAPK cascade genes in Brassica rapa and plant phylogenetics. Hortic Res England. 2017;4(1):17079.
He Y, Meng X. MAPK signaling: emerging roles in lateral root formation. Trends Plant Sci. 2020;25(2):126–9.
Xu R, Duan P, Yu H, Zhou Z, Zhang B, Wang R, Li J, Zhang G, Zhuang S, Lyu J, et al. Control of grain size and weight by the OsMKKK10-OsMKK4-OsMAPK6 signaling pathway in Rice. Mol Plant. 2018;11(6):860–73.
Li H, Ding Y, Shi Y, Zhang X, Zhang S, Gong Z, Yang S. MPK3- and MPK6-mediated ICE1 phosphorylation negatively regulates ICE1 stability and freezing tolerance in Arabidopsis. Dev Cell. 2017;43(5):630–42.
Zhang X, Guan H, Dai Z, Guo J, Shen Y, Cui G, Gao W, Huang L. Functional analysis of the Isopentenyl Diphosphate Isomerase of Salvia miltiorrhiza via color complementation and RNA interference. MOLECULES. 2015;20(11):20206–18.
Bai Z, Li W, Jia Y, Yue Z, Jiao J, Huang W, Xia P, Liang Z. The ethylene response factor SmERF6 co-regulates the transcription of SmCPS1 and SmKSL1 and is involved in tanshinone biosynthesis in Salvia miltiorrhiza hairy roots. PLANTA. 2018;248(1):243–55.
Ding K, Pei T, Bai Z, Jia Y, Ma P, Liang Z. SmMYB36, a Novel R2R3-MYB Transcription Factor, Enhances Tanshinone Accumulation and Decreases Phenolic Acid Content in Salvia miltiorrhiza Hairy Roots. Sci Rep Uk. 2017;7(1):5104.
Cao W, Wang Y, Shi M, Hao X, Zhao W, Wang Y, Ren J, Kai G. Transcription factor SmWRKY1 positively promotes the biosynthesis of Tanshinones in Salvia miltiorrhiza. Front Plant Sci. 2018;9:554.
Jia Y, Bai Z, Pei T, Ding K, Liang Z, Gong Y. The Protein Kinase SmSnRK2.6 Positively Regulates Phenolic Acid Biosynthesis in Salvia miltiorrhiza by Interacting with SmAREB1. Front Plant Sci. 2017;8:1384.
Yang N, Zhou W, Su J, Wang X, Li L, Wang L, Cao X, Wang Z. Overexpression of SmMYC2 increases the production of phenolic acids in Salvia miltiorrhiza. Front Plant Sci. 2017;8:1804.
Sethi V, Raghuram B, Sinha AK, Chattopadhyay S. A mitogen-activated protein kinase Cascade module, MKK3-MPK6 and MYC2, is involved in blue light-mediated seedling development inArabidopsis. Plant Cell. 2014;26(8):3343–57.
Li X, Guo H, Qi Y, Liu H, Zhang X, Ma P, Liang Z, Dong J. Salicylic acid-induced cytosolic acidification increases the accumulation of phenolic acids in Salvia miltiorrhiza cells. Plant Cell Tissue Organ Culture (PCTOC). 2016;126(2):333–41.
Xiao Y, Gao S, Di P, Chen J, Chen W, Zhang L. Methyl jasmonate dramatically enhances the accumulation of phenolic acids in Salvia miltiorrhiza hairy root cultures. Physiol Plant. 2009;137(1):1–9.
Liang Z, Ma Y, Xu T, Cui B, Liu Y, Guo Z, Yang D. Effects of abscisic acid, gibberellin, ethylene and their interactions on production of phenolic acids in salvia miltiorrhiza bunge hairy roots. PLoS One. 2013;8(9):e72806.
Li N, Han X, Feng D, Yuan D, Huang L. Signaling crosstalk between salicylic acid and ethylene/Jasmonate in plant defense: do we understand what they are whispering? Int J Mol Sci. 2019;20(3):671.
Burger M, Chory J. Stressed out about hormones: how plants orchestrate immunity. Cell Host Microbe. 2019;26(2):163–72.
Li S, Wu Y, Kuang J, Wang H, Du T, Huang Y, Zhang Y, Cao X, Wang Z. SmMYB111 is a key factor to phenolic acid biosynthesis and interacts with both SmTTG1 and SmbHLH51 inSalvia miltiorrhiza. J Agr Food Chem. 2018;66(30):8069–78.
Hao G, Jiang X, Feng L, Tao R, Li Y, Huang L. Cloning, molecular characterization and functional analysis of a putative R2R3-MYB transcription factor of the phenolic acid biosynthetic pathway in S. miltiorrhiza Bge. F. alba. Plant Cell Tissue Organ Culture (PCTOC). 2016;124(1):151–68.
Zhang S, Ma P, Yang D, Li W, Liang Z, Liu Y, Liu F. Cloning and characterization of a putative R2R3 MYB transcriptional repressor of the Rosmarinic acid biosynthetic pathway from Salvia miltiorrhiza. PLoS One. 2013;8(9):e73259.
Xu J, Li Y, Wang Y, Liu H, Lei L, Yang H, Liu G, Ren D. Activation of MAPK kinase 9 induces ethylene and Camalexin biosynthesis and enhances sensitivity to salt stress inArabidopsis. J Biol Chem. 2008;283(40):26996–7006.
Furlan G, Nakagami H, Eschen-Lippold L, Jiang X, Majovsky P, Kowarschik K, Hoehenwarter W, Lee J, Trujillo M. Changes in PUB22 Ubiquitination modes triggered by Mitogen-Activated Protein Kinase3 dampen the immune response. Plant Cell. 2017;29(4):726–45.
He Y, Xu J, Wang X, He X, Wang Y, Zhou J, Zhang S, Meng X. The Arabidopsis pleiotropic drug resistance transporters PEN3 and PDR12 mediate Camalexin secretion for resistance toBotrytis cinerea. Plant Cell. 2019;31(9):2206–22.
Park J, Kim TH, Takahashi Y, Schwab R, Dressano K, Stephan AB, Ceciliato PHO, Ramirez E, Garin V, Huffaker A, et al. Chemical genetic identification of a lectin receptor kinase that transduces immune responses and interferes with abscisic acid signaling. Plant J. 2019;98(3):492–510.
Zhao J, Chen Q, Zhou S, Sun Y, Li X, Li Y. H2Bub1 RegulatesRbohD-dependent hydrogen peroxide signal pathway in the defense responses toVerticillium dahliae toxins. Plant Physiol. 2020;182(1):640–57.
Long L, Xu F, Zhao J, Li B, Xu L, Gao W. GbMPK3 overexpression increases cotton sensitivity to Verticillium dahliae by regulating salicylic acid signaling. Plant Sci. 2020;292:110374.
Teardo E, Carraretto L, Moscatiello R, Cortese E, Vicario M, Festa M, Maso L, De Bortoli S, Calì T, Vothknecht UC, et al. A chloroplast-localized mitochondrial calcium uniporter transduces osmotic stress in Arabidopsis. Nat Plants. 2019;5(6):581–8.
Takahashi F, Yoshida R, Ichimura K, Mizoguchi T, Seo S, Yonezawa M, Maruyama K, Yamaguchi-Shinozaki K, Shinozaki K. The mitogen-activated protein kinase Cascade MKK3–MPK6 is an important part of the Jasmonate signal transduction pathway inArabidopsis. Plant Cell. 2007;19(3):805–18.
Su J, Zhang M, Zhang L, Sun T, Liu Y, Lukowitz W, Xu J, Zhang S. Regulation of Stomatal immunity by interdependent functions of a pathogen-responsive MPK3/MPK6 Cascade and Abscisic acid. Plant Cell. 2017;29(3):526–42.
Zhu Q, Shao Y, Ge S, Zhang M, Zhang T, Hu X, Liu Y, Walker J, Zhang S, Xu J. A MAPK cascade downstream of IDA-HAE/HSL2 ligand-receptor pair in lateral root emergence. Nat Plants. 2019;5(4):414–23.
Asai T, Tena G, Plotnikova J, Willmann MR, Chiu W, Gomez-Gomez L, Boller T, Ausubel FM, Sheen J. MAP kinase signalling cascade in Arabidopsis innate immunity. Nature (London). 2002;415(6875):977–83.
Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39(suppl):W29–37.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–80.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4.
Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406–25.
Gasteiger E. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31(13):3784–8.
Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. Meme suite: tools for motif discovery and searching. Nucleic Acids Res. 2009;37(Web Server):W202–8.
Li Y, Chen Y, Zhou L, You S, Deng H, Chen Y, Alseekh S, Yuan Y, Fu R, Zhang Z, et al. MicroTom metabolic network: rewiring tomato metabolic regulatory network throughout the growth cycle. Mol Plant. 2020;13(8):1203–18.
Zhou W, Huang Q, Wu X, Zhou Z, Ding M, Shi M, Huang F, Li S, Wang Y, Kai G. Comprehensive transcriptome profiling of Salvia miltiorrhiza for discovery of genes associated with the biosynthesis of tanshinones and phenolic acids. Sci Rep Uk. 2017;7(1):10554.
Acknowledgements
We greatly appreciate Professor Min Duan and Dr. Ningjuan Fan from the Biochemistry and Molecular Platform of the College of Life Sciences, Northwest A&F University, for their help in providing experimental instruments and instructions.
Funding
The transcriptomic data analyses in this study were financially supported by the National Natural Science Foundation of China (31670301 and 31670295), and the experimental material was supported by the Open Fund of the Shanghai Key Laboratory of Plant Functional Genomics and Resources (PFGR201903) and the Natural Science Basic Research Plan in Shaanxi Province in China (2019JM-149). The funding bodies played no role in not only the design of the study but also the collection and analysis of data and manuscript writing.
Author information
Authors and Affiliations
Contributions
JED and PDM conceived the project and reviewed the manuscript. YFX and MLD collected the public dataset and executed the bioinformatics analysis. BZ contributed to bioinformatics analysis and the making of all the figures and tables. YFX, TLP and JY performed the experiments. YFX drafted the manuscript. All of the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Ethical approval and consent are not applicable to this research.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1 :Table S1.
Sequences belonging to the SmMAPK family. Table S2. Summary data of the set of genes belonging to the SmMAPK family. Table S3. The content of cis-acting elements present in the promoter sequences associated with genes belonging to the SmMAPK family. Table S4. Enzymes participating in the synthesis of phenolic acids and tanshinones in S. miltiorrhiza. Table S5. Transcription factors regulating the synthesis of phenolic acids and tanshinones in S. miltiorrhiza. Table S6. The query sequences from A. thaliana. Table S7. Primers used for qRT-PCR and vector construction.
Additional file 2 :Figure S1.
Conserved motifs in the products of genes belonging to the SmMAPK family, as predicted by MEME software. Figure S2. A proposed interaction network involving the products of genes belonging to the SmMAPK family. Figure S3. Pearson correlation coefficient of R value between the Ct value of the qRT-PCR results and the log2 RPKM values from the RNA-seq analysis. Pearson correlation coefficient of R values were visualized by TBtools.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xie, Y., Ding, M., Zhang, B. et al. Genome-wide characterization and expression profiling of MAPK cascade genes in Salvia miltiorrhiza reveals the function of SmMAPK3 and SmMAPK1 in secondary metabolism. BMC Genomics 21, 630 (2020). https://doi.org/10.1186/s12864-020-07023-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-020-07023-w