
Abstract
Background
Trypanosomatids of the genus Leishmania are parasites of mammals or reptiles transmitted by bloodsucking dipterans. Many species of these flagellates cause important human diseases with clinical symptoms ranging from skin sores to life-threatening damage of visceral organs. The genus Leishmania contains four subgenera: Leishmania, Sauroleishmania, Viannia, and Mundinia. The last subgenus has been established recently and remains understudied, although Mundinia contains human-infecting species. In addition, it is interesting from the evolutionary viewpoint, representing the earliest branch within the genus and possibly with a different type of vector. Here we analyzed the genomes of L. (M.) martiniquensis, L. (M.) enriettii and L. (M.) macropodum to better understand the biology and evolution of these parasites.
Results
All three genomes analyzed were approximately of the same size (~ 30 Mb) and similar to that of L. (Sauroleishmania) tarentolae, but smaller than those of the members of subgenera Leishmania and Viannia, or the genus Endotrypanum (~ 32 Mb). This difference was explained by domination of gene losses over gains and contractions over expansions at the Mundinia node, although only a few of these genes could be identified. The analysis predicts significant changes in the Mundinia cell surface architecture, with the most important ones relating to losses of LPG-modifying side chain galactosyltransferases and arabinosyltransferases, as well as β-amastins. Among other important changes were gene family contractions for the oxygen-sensing adenylate cyclases and FYVE zinc finger-containing proteins.
Conclusions
We suggest that adaptation of Mundinia to different vectors and hosts has led to alternative host-parasite relationships and, thereby, made some proteins redundant. Thus, the evolution of genomes in the genus Leishmania and, in particular, in the subgenus Mundinia was mainly shaped by host (or vector) switches.
Similar content being viewed by others


Background
Obligate flagellate parasites of the family Trypanosomatidae infect insects, leeches, vertebrates, and plants [1,2,3]. They have one (monoxenous species) or two hosts (dixenous species) in their life cycle [4,5,6]. Dixenous representatives belong to the genera Endotrypanum, Leishmania, Paraleishmania, Phytomonas, and Trypanosoma and some of them are of medical and/or economic importance [7,8,9]. It is generally accepted that all dixenous trypanosomatids have originated from their monoxenous kin [10]. Supporting this, in the current taxonomical system, the dixenous genera Endotrypanum, Leishmania, Paraleishmania are united with the monoxenous genera Borovskyia, Crithidia, Leptomonas, Lotmaria, Novymonas, and Zelonia into the subfamily Leishmaniinae [11, 12], while the dixenous genus Phytomonas is included into subfamily Phytomonadinae along with the monoxenous genera Herpetomonas and Lafontella [13].
Parasites of the genus Leishmania infect mammals or reptiles and cause various diseases named leishmaniases. For humans, this translates into over 350 million people being at risk of infection primarily in the tropical and subtropical regions [14]. These parasites are transmitted by bloodsucking phlebotomine sand flies (Psychodidae) or, possibly, biting midges (Ceratopogonidae) [15, 16] and manifest the infection by a range of clinical symptoms from innocuous skin lesions to fatal visceral organ failures [7].
Currently, the following four subgenera are recognized within the genus Leishmania. These are Leishmania (Leishmania), L. (Mundinia), L. (Sauroleishmania), and L. (Viannia) [17]. They are not only well-defined phylogenetically, but can also be delineated by host specificity or clinical picture. The most enigmatic of them is Mundinia [18], the last established subgenus [17], which, as of now, contains only four described species: L. enriettii, L. macropodum, L. martiniquensis, and L. orientalis [19,20,21,22]. In addition, there are isolates from Ghana, likely representing a separate species, which is phylogenetically close to L. orientalis [20].
Leishmania (Mundinia) spp. are of special interest for, at least, four main reasons. Firstly, in this group, human pathogens – L. (M.) orientalis, L. (M.) martiniquensis and parasites from Ghana – are intermingled with species non-pathogenic to humans, namely L. (M.) enriettii and L. (M.) macropodum [20, 23]. Leishmania (M.) enriettii infects guinea pigs in South America [24, 25], while L. (M.) macropodum was found in Australian macropods [26, 27]. In addition, parasites apparently belonging to L. martiniquensis have been also recorded in cows and horses [28,29,30]. Secondly, a significant portion of human patients infected with Leishmania (Mundinia) are immunocompromised [31,32,33], indicating that these parasites may actively explore new developmental niches [10, 34]. A similar situation has been documented in some thermo-tolerant monoxenous trypanosomatids [35,36,37]. Thirdly, Mundinia spp. may be transmitted primarily not by phlebotomine sand flies of the genera Phlebotomus and Lutzomyia as for other leishmaniae, but by biting midges or other genera of sand flies, although more work is needed to confirm this with certainty [15, 38]. Fourthly, and finally, in all phylogenetic reconstructions, L. (Mundinia) represents the earliest branch within the genus Leishmania, suggesting its ancient origin prior to the breakup of Gondwana [2, 39].
For all these reasons, members of the subgenus Mundinia qualify as crucial for comparative genomic analyses, as they may shed light on the evolution of Leishmania and its pathogenicity for humans. Similar analyses have been done and reported for L. (Sauroleishmania) [40, 41], L. (Viannia) [42,43,44,45], L. (Leishmania) [46, 47], leaving Mundinia understudied in this respect.
In this work, we sequenced and analyzed genomes of three Leishmania (Mundinia) species, which represent the major clades of the subgenus: L. (M.) enriettii MCAV/BR/1945/LV90 originating from southern Brazil, L. (M.) macropodum MMAC/AU/2004/AM-2004 originating from northern Australia, and L. (M.) martiniquensis MHOM/MQ/1992/MAR1 originating from the Caribbean island of Martinique. The genomic sequence of L. (M.) enriettii MCAV/BR/1945/LV90 complemented a previously obtained one, which belongs to a different isolate of the same species (MCAV/BR/1995/CUR3) and is available from the TriTryp database.
Methods
Origin of isolates, cultivation, amplification, sequencing and species verification
Promastigotes were cultured in the M199 medium (Sigma−Aldrich, St. Louis, MO, United States) containing 10% heat-inactivated fetal bovine calf serum (FBS; Thermo Fisher Scientific, Waltham, MA, United States), supplemented with 1% Basal Medium Eagle vitamins (Sigma−Aldrich), 2% sterile urine and 250 μg/ml of amikacin (Bristol-Myers Squibb, New York, NY, United States).
Total genomic DNA was isolated from 10 ml of trypanosomatid cultures with the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. 18S rRNA gene was amplified using primers S762 and S763 [48], following the previously described protocol [13]. These PCR fragments were sequenced directly at Macrogen Europe (Amsterdam, Netherlands) as described previously [49]. The identity of species under study was confirmed by BLAST analysis [50].
Whole-genome and whole-transcriptome sequencing and analysis
The genomes and whole transcriptomes of Leishmania (Mundinia) isolates were sequenced as described previously [35, 51, 52] using the Illumina HiSeq and NovaSeq technologies with TruSeq adapters for the libraries preparation, respectively, at Macrogen Inc. (Seoul, South Korea). 43 and 47 million 100 nt paired-end raw reads on average were produced for genomes and transcriptomes, respectively (see statistics below). The genome completeness and annotation quality were assessed using BUSCO software [53]. The raw reads were trimmed with Trimmomatic v. 0.32 [54] with the following settings: ILLUMINACLIP:TruSeq3-PE-2.fa:2:20:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:75, quality-checked with FASTQC program v.0.11.5, and then assembled de novo with the Spades Genome assembler v. 3.10.1 with the default settings and automatic k-mer selection (k-mers of 21, 33 and 55 were used) [55]. The Trinity assembler v. 2.4.0 [56] was used to reconstruct the transcriptomes de novo with the minimal contig length of 150. Resulting genome assemblies were investigated for potential contamination using the BlobTools software implementing Bowtie2 [57] for genome read mapping and Hisat2 for transcriptome read mapping [58], both with the default settings. Only those read pairs were used where at least one read was present in some contig with the transcriptome read coverage higher than 10 or in a contig with Leishmania, Leptomonas, or Trypanosoma term in first 100 best Diamond hits. Other read pairs were filtered out (Additional file 1: Figure S1, Additional file 2: Figure S2, Additional file 3: Figure S3, Additional file 4: Figure S4, Additional file 5: Figure S5, Additional file 6: Figure S6). Resulting assemblies (CovPlots, Additional file 7: Figure S7, Additional file 8: Figure S8, Additional file 9: Figure S9) were further inspected and curated manually. Parameters of the genome assemblies were estimated using QUAST v. 4.5 [59]. Raw reads were submitted to NCBI SRA under accession numbers SRX5006814, SRX5006815, and SRX5006816 (Bioproject: PRJNA505413) for L. (M.) enriettii MCAV/BR/1945/LV90, L. (M.) macropodum MMAC/AU/2004/AM-2004, and L. (M.) martiniquensis MHOM/MQ/1992/MAR1, respectively.
Genome annotation was performed with the Companion software [60] using transcriptome evidence, Leishmania major as a reference organism, and pseudochromosome contiguation with default settings. Transcriptome evidence was generated with the Cufflinks, mapping was performed with the Hisat2 with --dta-cufflinks parameter [58].
Synteny analysis
Synteny analysis was performed using SyMAP v. 4.2 [61] with the following settings: minimum size of sequence to load, 500 bp; minimum number of anchors required to define a synteny block, 7; synteny blocks were merged in case of overlaps, and only the larger block was kept if two synteny blocks overlapped on a chromosome. In case of Leishmania (Mundinia) genomes sequenced in this study, pseudochromosome level assembly built using Companion software with L. major Friedlin genome as a reference was used for the analysis instead of scaffolds in order to reduce computational time.
Genome coverage analysis and ploidy estimation
Per-base read coverage was calculated for fifty longest scaffolds and all pseudochromosome level sequences using BEDTools v. 2.26.0 genomecov tool [62] on the read mappings generated with Bowtie2 as described above. Mean genome and scaffold/pseudochromosme coverage was calculated using a custom Python script. Ploidy was estimated based on relative coverage values: mean coverage for each of the fifty longest scaffolds and all psedochoromosome level sequences was divided by mean genome coverage and ploidy was inferred under the assumption that the majority of chromosomes are diploid. Coverage plots for 50 longest scaffolds were generated using weeSAM tool v. 1.5 (http://bioinformatics.cvr.ac.uk/blog/weesam-version-1-5/).
Variant calling
Prior to variant calling, duplicates removal and local re-alignment were performed on the respective read mappings using GATK v. 4.1.2.0 MarkDuplicates and IndelRealigner tools with the following parameter differing from the default: --REMOVE_DUPLICATES = true [63]. Variant calling was performed using Platypus v. 0.1.5 [64] with the default settings and only SNPs were considered in further analyses.
Inference of protein orthologous groups and phylogenomic analyses
Analysis of protein orthologous groups was performed on a dataset containing 41 trypanosomatid species (including four representatives of the subgenus Mundinia, Additional file 16: Table S1) and a eubodonid Bodo saltans as an outgroup, using OrthoFinder v. 1.1.8 with the default settings [65]. Out of a total 551 OGs containing only one protein for each species, 92 were selected for the phylogenomic inference according to the following criteria: i) average percent identity within the group ≥60%; ii) maximum percentage of gaps per sequence in the alignment before trimming – 40%; iii) maximum percentage of gaps per sequence in the alignment after trimming – 10%. The amino acid sequences of each gene were aligned using Muscle v. 3.8.31 [66]. The average percent identity within each OG was calculated using the alistat script from the HMMER package v.3.1 [67]. The alignments were trimmed using trimAl v. 1.4.rev22 with the “-strict” option [68]. The resulting concatenated alignment contained 32,460 columns. The maximum likelihood tree was inferred in IQ-TREE v. 1.6.3 with the JTT + F + I + G4 model and 1000 bootstrap replicates [69, 70]. For the construction of the Bayesian tree PhyloBayes-MPI 1.7b was run for over 9000 iterations under the GTR-CAT model with four discrete gamma categories [71]. Every second tree was sampled and first 25% of them were discarded as “burn-in”. The final tree was visualized using FigTree v.1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/). Gains/losses and expansions/contractions of protein families were analyzed using the COUNT software with Dollo’s and Wagner’s (gain penalty set to 3) parsimony algorithms, respectively [72]. For gene ontology (GO) annotation of gene families gained/lost/expanded/contracted at certain nodes Blast2GO Basic software [73] was used with the maximum number of BLAST hits set to 10 and other settings left as default. Assignment of KEGG IDs to the proteins of interest was performed via BlastKOALA server with a target database of eukaryotes and prokaryotes at the family and genus levels, respectively [74]. The analysis of OGs shared among Leishmania was performed using UpSetR package [75].
Analysis of amastin repertoire
Amastin sequences of L. major Friedlin, Trypanosoma brucei TREU927, and Trypanosoma cruzi CL Brener Esmeraldo were downloaded from the TriTrypDB release 41 and used as queries in BLAST search with an E-value threshold of 10− 20 against a database of annotated proteins of Crithidia fasciculata, Endotrypanum monterogeii, Leishmania braziliensis MHOM/BR/75/M2904, Leishmania (Mundinia) spp., Leptomonas pyrrhocoris H10, and Trypanosoma grayi ANR4. The resulting sequences were aligned using Muscle v.3.8.31 with the default parameters [66]. P-distances were calculated using MEGA 7 software [76], and the hits with p-distance to the α-amastin of T. brucei (Additional file 17: Table S2) exceeding 0.9 and query coverage < 50% were excluded from further analyses. The resulting alignment was trimmed using TrimAl v.1.4.rev22 with the ‘-gappyout’ option [68]. Maximum-likelihood phylogenetic tree was inferred on the final dataset containing 384 sequences and 436 characters using IQ-TREE v.1.5.3 with the VT + F + G4 model and 1000 bootstrap replicates [69, 70].
Analysis of side chain galactosyltransferases
The identification of the side chain galactosyltransferases (SCGs) was performed as described previously [77]. Proteins with p-distances to SCGs of L. major exceeding 0.8 were excluded from further analysis (Additional file 18: Table S3 and Additional file 19: Table S4). Phylogenetic reconstruction was performed using IQ-TREE v.1.5.3 with 1000 bootstrap replicates and VT + F + I + G4 and JTT + F + G4 models for the SCGs and side chain arabinosyltransferases (SCAs), respectively.
Analyses of other proteins within OGs gained/lost at certain nodes
For the identification of putative phosphatydylinositol glycan class Y proteins (PIG-Y), we have performed sensitive homology searches using the HMMER package v.3.1 [67] and a model build using aligned sequences of trypanosomatid annotated as PIG-Y from the TriTrypDB release 41 [78]. Phylogenetic analysis of PIG-Y was performed similarly to amastins, with the JTT + I + G4 model as best-fitting and excluding sequences with p-distances to the reference set higher than 0.8 (Additional file 20: Table S5). The analysis of ferrochelatase sequences was performed similarly (Additional file 21: Table S6), with the JTT + I + G4 phylogenetic model.
Results
Assembly and annotation of three Leishmania (Mundinia) genomes
The three sequenced genomes were assembled and annotated, yielding total lengths of 29.95, 29.59, and 29.83 Mbp for L. (M.) martiniquensis MHOM/MQ/1992/MAR1, L. (M.) macropodum MMAC/AU/2004/AM-2004, and L. (M.) enriettii MCAV/BR/1945/LV90, respectively for the scaffolds longer than 500 bp (Additional file 22: Table S7). The N50 values and largest scaffold sizes varied from 24.17 to 33.45 kbp, and from 181 to 225 kbp for L. (M.) enriettii and L. (M.) martiniquensis, respectively. Genomic reads coverage analysis (Additional file 10: Figure S10) indicates that coverage is fairly uniform across Mundinia genome assemblies, with the regions of coverage close to median values (exceeding 40x but lower than 150x) combined together accounting for ~ 91, 89 and 80% of genome assembly length for L. (M.) martiniquensis, L. (M.) macropodum, and L. (M.) enriettii, respectively. The results of variant calling suggest that the genome of L. (M.) enriettii carrying 12,379 SNPs is characterized by higher variation levels than those of L. (M.) martiniquensis and L. (M.) macropodum with 1765 and 4834 identified SNPs, respectively (Additional file 22: Table S7). The number of homozygous SNPs identified in L. (M.) martiniquensis, L. (M.) macropodum, and L. (M.) enriettii genome assemblies were as low as 64, 67 and 121, respectively, suggesting minimal number of misassembly events (Additional file 22: Table S7).
Expectedly, the results of ploidy analysis suggest that Leishmania (Mundinia) spp. demonstrate variable degree of aneuploidy (Additional file 23: Table S8). In L. (M.) martiniquensis all pseudochromosome level sequences appear to be diploid, except for chromosome 31. The genome of L. (M.) enriettii displays the highest level of aneuploidy among the analyzed species, with nine chromosomes of variable ploidy levels (Additional file 23: Table S8).
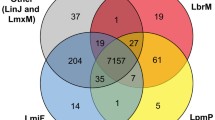
All the analyzed genomes are predicted to encode around 8000 genes and had complete BUSCOs percentage of around 72% (Additional file 22: Table S7). For comparison, the previously sequenced genome of another isolate of L. (M.) enriettii – MCAV/BR/1995/CUR3 (LEM3045) – has similar, albeit slightly larger (partially due to a ~ 60-fold higher gap content), size of 30.9 Mbp (29.2 Mbp in 36 scaffolds) and was predicted to encode 8831 genes. Mundinia genomes obtained in this study show high degree of synteny to publicly available ones and the assembly for L. major Friedlin (Additional file 11: Figure S11). From 93 to 98% of genes identified in the assemblies obtained in this study are located within synteny blocks in various intra- and interspecies comparisons (Additional file 11: Figure S11, panel B). The absence of collapsed repeats and highly similar genes in the obtained assemblies is supported by the absence of regions of double coverage (i.e., regions covered by two or more synteny blocks) as compared to publicly available genomes (Additional file 11: Figure S11, panel B). Annotated proteins of all representatives of the genus Leishmania within our dataset cluster into 8657 OGs. Most of these groups (83%, 7175 OGs) are shared among all four subgenera (Fig. 1). Mundinia spp. appear to possess the lowest number of the subgenus-specific OGs (~ 100), while the representatives of L. (Leishmania) have ~ 500 such groups.

The phyletic patterns for OGs identified in four subgenera of the genus Leishmania: L. (Leishmania), L. (Viannia), L. (Mundinia), and L. (Sauroleishmania). An UpSetR plot shows the numbers of orthologous groups uniquely shared among four subgenera of Leishmania. Intersection size (the number of shared OGs) is plotted on Y axis; dataset intersection options are indicated on the X axis with black circles
Phylogenomic analysis
The Maximum Likelihood and Bayesian trees inferred using the matrix of 92 single-copy OGs displayed identical topologies with almost all branches having maximal bootstrap percentage and posterior probabilities (except for two modestly resolved branches of monoxenous Leishmaniinae: Lotmaria passim and intermingled species of the Leptomonas – Crithidia clade). Our results confirmed the phylogenetic position of Mundinia as the earliest branch within the genus Leishmania (Fig. 2), which has been inferred in previous studies [2, 39]. It is also in agreement with the recently published phylogenetic trees of Mundinia spp., which were reconstructed using several single phylogenetic markers [20, 23].

The phylogenetic tree of trypanosomatids and Bodo saltans based on the alignment of 92 conserved proteins. Only bootstrap support values lower than 100% and posterior probabilities lower than 1 are shown. The scale bar represents 0.05 substitutions per site. Pie charts depict relative proportions of OGs gains/losses and expansions/contractions in green/red and blue/magenta colors, respectively. The area of the pie charts is proportional to a total number of OGs gained/lost or expanded/contracted at a certain node. The nodes corresponding to the subgenus Mundinia and to the all other Leishmania are highlighted in orange and cyan, respectively
Gene gains and losses at the Leishmania (Mundinia) node
The Leishmania (Mundinia) node was heavily dominated by gene losses. There were 13 gained and 234 lost OGs at this node (Fig. 2, Additional file 24: Table S9). All 13 gained and 148 lost OGs contained genes encoding hypothetical proteins. In contrast, the node uniting the three remaining subgenera was dominated by gene gains with 79 gained (71 OGs contained genes encoding hypothetical proteins) and 34 lost (22 OGs contained genes encoding hypothetical proteins) (Fig. 2, Additional file 25: Table S10).
The annotations for sequences within OGs lost at the L. (Mundinia) node indicate changes in the surface architecture of the parasites of this subgenus, exemplified by the losses of putative amastins, glycosylphosphatidylinositol (GPI) anchor biosynthesis and turnover proteins. Amastins are a large family of surface glycoproteins, highly expressed in the amastigote stage of several trypanosomatids, such as T. cruzi and Leishmania spp. [79]. They are essential for establishing infection in macrophages [80, 81] and, therefore, are significantly reduced in lizard-parasitizing L. tarentolae, which cannot efficiently replicate in this type of cells and rarely forms amastigotes [41].
The results of our gene content evolution analyses suggest that three OGs containing putative amastins were lost at the L. (Mundinia) node (Additional file 24: Table S9). According to the phylogenetic analysis (Additional file 12: Figure S12), two of those OGs – OG0008773 and OG0009479 (Additional file 24: Table S9) – contain putative β-amastin-like proteins, homologues of which were lost in all analyzed Leishmania spp. except for L. major and L. braziliensis, respectively. OG0009537 incorporates γ-amastin-related proteins, identified in the genomes of the monoxenous Leishmaniinae, but lost in all L. (Leishmania) spp. [82]. Overall, 33, 19 and 23 amastin-like sequences were identified in L. (M.) martiniquensis, L. (M.) macropodum, and L. (M.) enriettii, respectively. L. (Mundinia) genomes encode representatives of all four amastin subfamilies, including Leishmania-specific δ-amastins.
The amastin polypeptides are linked to the parasite’s outer membrane via a GPI anchor [83, 84]. Two enzymes involved in GPI-anchor synthesis and GPI-anchored protein turnover, phosphatidylinositol N-acetylglucosaminyltransferase (subunit Y) and glycosylphosphatidylinositol phospholipase-C (GPI-PLC), respectively, also appear to be lost at the L. (Mundinia) node. However, a careful inspection of the results has shown that GPI-PLC is absent not only from Mundinia, but also from other subgenera of Leishmania, as well as from Endotrypanum. The only exception is L. panamensis with a partial sequence of unknown function returning a short hit to the GPI-PLC. This hit resulted in erroneous inference of the putative GPI-PLC presence at the L. (Leishmania) node by the Dollo’s parsimony algorithm. Putative GPI-PLC have been identified in all species within our dataset, except for dixenous Leishmaniinae, C. expoeki, and Phytomonas spp. In trypanosomatids, phosphatidylinositol N-acetyl-glucosaminyl-transferase, the enzyme catalyzing the first step of GPI biosynthesis, is composed of seven proteins: phosphatydyl-inositol glycan class A (PIG-A), PIG-C, PIG-H, PIG-Q, PIG-P, PIG-Y, and dolichyl-phosphate mannosyl-transferase polypeptide 2 (DPM2) [85]. All these proteins were identified in L. (Mundinia), with the exception of DMP2 and PIG-Y being absent from the genome of L. (M.) macropodum. The analysis of orthologous groups revealed that PIG-Y sequences fall into two different OGs, one of which appears to be absent in L. (Mundinia). More sensitive HMM-based searches led to the identification of PIG-Y proteins in several other trypanosomatids. The phylogenetic analysis confirmed the presence of two separate groups of PIG-Y sequences, only one of which contains L. (Mundinia) subunits (Additional file 13: Figure S13). Most of the L. (Leishmania) sequences fall into the latter group, while the representatives of the other clade appear to be in the process of pseudogenization in L. (Leishmania), as suggested by the presence of the identifiable pseudogenes in L. major and L. tarentolae.
We have also analyzed the repertoire of side chain galactosyltransferases (SCGs) and side chain arabinosyltransferases (SCAs), performing chemical modifications of the GPI-anchored lipophosphoglycan (LPG) on the cell surface of the Leishmaniinae [77, 86, 87], with the potential effect on host-parasite interactions [88,89,90]. The genome of L. (M.) martiniquensis encodes five SCGs, while those of L. (M.) macropodum and L. (M.) enriettii, sequenced in this study, contain four putative members of SCG/L/R family (Additional file 14: Figure S14). Thus, in L. (Mundinia) the number of SCG-encoding genes is substantially lower than in L. major, L. braziliensis and L. infantum, carrying 14, 17 and 12 genes, respectively. L. (Mundinia) SCG proteins cluster with those of L. braziliensis, and together they form a sister clade to the SCGs of L. major and L. infantum. In addition, L. (Mundinia) spp. contain sequences related to the SCGR1–6, while putative SCGL-encoding genes were not identified, similarly to the situation observed in L. braziliensis [91, 92]. Overall, the SCG/L/R repertoire in L. (Mundinia) is most similar to the one in L. braziliensis, with the exception of the SCG expansion in L. braziliensis, which is not documented in L. (Mundinia). In addition, L. (Mundinia) spp. possess SCA and SCA-like sequences, which are absent in L. braziliensis (Additional file 14. Figure S14).
A few genes encoding metabolic proteins appear to be lost in L. (Mundinia). An important enzyme of folate metabolism is methylene-tetrahydrofolate reductase (MTFR), which converts 5-methyltetrahydrofolate into 5,10-methylene-tetrahydrofolate and is required for the formation of activated C1 units used in the synthesis of both thymidylate by thymidylate synthase/dihydrofolate reductase and of methionine from cysteine by methionine synthase [93, 94]. MTFR is present in Bodo saltans, Paratrypanosoma confusum, Blechomonas alayai, and all Leishmaniinae with the sole exception of L. (Mundinia). In addition to this, it is also absent from trypanosomes and Phytomonas. However, the absence of MTFR does not imply auxotrophy for methionine, since all trypanosomatids seem to be able to synthesize this amino acid by an alternative route using homocysteine S-methyltransferase [95].
Following the observation that ferrochelatase (FeCH), the terminal enzyme in the heme biosynthetic pathway catalyzing the insertion of iron into protoporphyrin IX [96], was lost in Leishmania (Additional file 25. Table S10), we have checked the presence of other enzymes of this pathway. Some trypanosomatids (Trypanosoma and Kentomonas), have lost the heme biosynthetic pathway completely, while others retained genes encoding the last three enzymes (Leishmaniinae, Angomonas and Strigomonas), or only ferrochelatase (Phytomonas and Herpetomonas) [97,98,99,100,101]. Protoporphyrin IX, a substrate of FeCH, is synthesized by a subsequent action of coproporphyrinogen oxidase and protoporphyrinogen oxidase [102]. Both enzymes were readily identifiable in the genomes of L. (Mundinia) spp., except for L. (M.) macropodum. Sequences of FeCH clustered in two separate OGs, only one of which incorporates the proteins of all three L. (Mundinia) spp. (Additional file 15: Figure S15). The other OG contains only the sequences of B. ayalai, E. monterogeiii, Phytomonas spp., and monoxenous representatives of the subfamily Leishmaniinae. The phylogenetic analysis of FeCH (Additional file 15: Figure S15) suggests the presence of two divergent sequences encoding this protein in the genomes of trypanosomatids, which is in agreement with the results of previous studies concluding that there might have been two different FeCH LGT events from bacteria to kinetoplastids [99]. Indeed, the FeCH sequences of C. fasciculata, falling into two different clades, exhibit only ~ 22% identity, giving best BLAST hits outside the Euglenozoa to the γ-proteobacterial sequences.
Kinetoplastids lack the capacity of de novo lysine biosynthesis. However, B. saltans, Leptomonas and Crithidia spp. use the enzyme diaminopimelate epimerase (DAP) to convert diaminopimelate, an amino acid present in the cell walls of gram-negative bacteria, into lysine [97]. In all other trypanosomatids, including L. (Mundinia), DAP has been lost. The loss of genes encoding this enzyme suggests that most of the trypanosomatids have lost their dependency on bacterial diaminopimelate and, thus, are lysine auxotrophs. Interestingly, the genomes of most L. (Leishmania) spp. still possess easily identifiable diaminopimelate epimerase pseudogenes, while no remnants of DAP-encoding genes could be found in other trypanosomatid genomes. This suggests that these genes could have been acquired by the common ancestor of all Leishmaniinae and then independently lost in different lineages of its dixenous descendants.
Gene family expansions and contractions at the Leishmania (Mundinia) node
In L. (Mundinia), 9 gene families were expanded (3 genes encoding hypothetical proteins) and 40 contracted (7 genes encoding hypothetical proteins) (Fig. 2; Additional file 26: Table S11), while in other subgenera, 11 gene families were expanded (4 genes encoding hypothetical proteins) and 7 contracted (3 genes encoding hypothetical proteins) (Fig. 2; Additional file 27: Table S12). The degree of gene family expansion/contraction is rather moderate, with the family size changes involving from 1 to 5 gene copies (Additional file 26: Table S11, Additional file 27: Table S12).
Oxygen-sensing adenylate cyclases (OG0000628) govern O2-dependent cAMP signaling via protein kinase A, and, consequently, cell survival and proliferation of Leishmania promastigotes under low concentration of oxygen [103]. Contraction of this gene family in L. (Mundinia) suggests that these parasites either rely on different mechanisms to deal with hypoxia or are under different environmental cues during development in their vectors.
Another interesting example is a contracted gene family encoding FYVE zinc finger-containing proteins (OG0001095). In eukaryotes, the FYVE domain is responsible for the recruitment of proteins to different organelles such as multivesicular bodies, endosomes, or phagosomes [104]. Membrane recruitment is mediated by the binding of the FYVE domain to membrane-embedded phosphatidylinositol-3-phosphate [105]. Why this gene family is contracted in L. (Mundinia) remains to be investigated further.
Discussion
The genomes of the three species of Leishmania (Mundinia) analyzed here are similar in size to that of L. (Sauroleishmania) tarentolae (~ 30 Mb), but smaller than those of the representatives of the subgenera L. (Leishmania) and L. (Viannia), as well as the genus Endotrypanum (~ 32 Mb). This correlates not only with the intuitively understandable domination of gene losses over gains and contractions over expansions, but also with the fact that both Mundinia and Sauroleishmania had switched to the new hosts or vectors. The majority of dixenous Leishmaniinae (i.e. Leishmania, Paraleishmania and Endotrypanum) parasitize mammals and are transmitted by phlebotomine sand flies and this, therefore, is the most likely ancestral variant of the life cycle. Meanwhile, Sauroleishmania spp. switched their vertebrate host from mammals to reptiles, whereas Mundinia spp. have substituted the phlebotomine sand fly hosts with biting midges and/or non-conventional sand flies. We speculate that adaptation to the new hosts or vectors has led to different, possibly simplified, host-parasite relationships and, thereby, made some of the previously used proteins redundant. Indeed, Sauroleishmania spp. demonstrate less specific relationships with their vertebrate hosts as compared to other Leishmania spp. Their promastigotes usually reside in the intestine or in the bloodstream, while occasionally formed amastigotes do not survive in macrophages [106].
Little is known about the relationships of L. (Mundinia) spp. and their vectors. However, our finding of a significant shrinkage of repertoires of the SCGs and SCAs in Mundinia, which are involved in interactions of promastigotes with the insect gut, implies simplification of the host-parasite relationships. At the same time, amastins and PIG-Y, which are primarily important for the survival of amastigotes in macrophages, showed generally the same evolutionary trends as in L. (Leishmania) and L. (Viannia), i.e. underwent independent losses. Moreover, those were mainly β-amastins, which are expressed in the vectorial part of the life cycle in T. cruzi [79]. In contrast, Sauroleishmania lost all amastigote-specific δ-amastins [41], whereas all other Leishmania subgenera preserved them.
In summary, we propose that the evolution of genomes in the genus Leishmania and, in particular, in the subgenus Mundinia was mainly shaped by host (or vector) switches.
Conclusions
In this work we have sequenced and analyzed genomes of several representatives of the most understudied Leishmania subgenus, Mundinia. Comparative analyses allowed us to gain additional insights into the origin of pathogenic Leishmania. We propose that the evolution of this genus was mainly driven by the host (or vector) switches.
Availability of data and materials
The datasets generated and analyzed during the current study will be available in the NCBI SRA repository under accession numbers SRX5006814, SRX5006815, and SRX5006816 (Bioproject: PRJNA505413) upon publication, https://www.ncbi.nlm.nih.gov/sra
Abbreviations
- GPI:
-
Glycosylphosphatidylinositol
- OG:
-
Orthogroup
- PIG-Y:
-
Phosphatydylinositol glycan class Y protein
- SCA:
-
Side chain arabinosyltransferase
- SCG:
-
Side chain galactosyltransferases
References
Maslov DA, Opperdoes FR, Kostygov AY, Hashimi H, Lukeš J, Yurchenko V. Recent advances in trypanosomatid research: genome organization, expression, metabolism, taxonomy and evolution. Parasitology. 2019;146(1):1–27.
Lukeš J, Butenko A, Hashimi H, Maslov DA, Votýpka J, Yurchenko V. Trypanosomatids are much more than just trypanosomes: clues from the expanded family tree. Trends Parasitol. 2018;34(6):466–80.
Vickerman K. Comparative cell biology of the kinetoplastid flagellates. In: Vickerman K, Preston TM, editors. Biology of Kinetoplastida, vol. 1. London: Academic; 1976. p. 35–130.
Maslov DA, Votýpka J, Yurchenko V, Lukeš J. Diversity and phylogeny of insect trypanosomatids: all that is hidden shall be revealed. Trends Parasitol. 2013;29(1):43–52.
McGhee RB, Cosgrove WB. Biology and physiology of the lower Trypanosomatidae. Microbiol Rev. 1980;44(1):140–73.
Podlipaev SA. Catalogue of world fauna of Trypanosomatidae (protozoa), vol. 144. Leningrad: Zoologicheskii Institut AN SSSR; 1990.
Bruschi F, Gradoni L. The leishmaniases: old neglected tropical diseases. Cham: Springer; 2018.
Stevens JR, Gibson WC. The evolution of pathogenic trypanosomes. Cad Saude Publica. 1999;15(4):673–84.
Camargo EP. Phytomonas and other trypanosomatid parasites of plants and fruit. Adv Parasitol. 1999;42:29–112.
Lukeš J, Skalický T, Týč J, Votýpka J, Yurchenko V. Evolution of parasitism in kinetoplastid flagellates. Mol Biochem Parasitol. 2014;195(2):115–22.
Jirků M, Yurchenko V, Lukeš J, Maslov DA. New species of insect trypanosomatids from Costa Rica and the proposal for a new subfamily within the Trypanosomatidae. J Eukaryot Microbiol. 2012;59(6):537–47.
Kostygov AY, Yurchenko V. Revised classification of the subfamily Leishmaniinae (Trypanosomatidae). Folia Parasitol. 2017;64:020.
Yurchenko V, Kostygov A, Havlová J, Grybchuk-Ieremenko A, Ševčíková T, Lukeš J, Ševčík J, Votýpka J. Diversity of trypanosomatids in cockroaches and the description of Herpetomonas tarakana sp. n. J Eukaryot Microbiol. 2016;63(2):198–209.
Alvar J, Velez ID, Bern C, Herrero M, Desjeux P, Cano J, Jannin J, den Boer M. Leishmaniasis worldwide and global estimates of its incidence. PLoS One. 2012;7(5):e35671.
Seblová V, Sádlová J, Vojtková B, Votýpka J, Carpenter S, Bates PA, Volf P. The biting midge Culicoides sonorensis (Diptera: Ceratopogonidae) is capable of developing late stage infections of Leishmania enriettii. PLoS Negl Trop Dis. 2015;9(9):e0004060.
Dvorák V, Shaw JJ, Volf P. Parasite biology: the vectors. In: Bruschi F, Gradoni L, editors. The leishmaniases: old neglected tropical diseases. Cham: Springer; 2018. p. 31–77.
Espinosa OA, Serrano MG, Camargo EP, Teixeira MM, Shaw JJ. An appraisal of the taxonomy and nomenclature of trypanosomatids presently classified as Leishmania and Endotrypanum. Parasitology. 2018;145(4):430–42.
Lainson R. On Leishmania enriettii and other enigmatic Leishmania species of the Neotropics. Mem Inst Oswaldo Cruz. 1997;92(3):377–87.
Muniz J, Medina H. Cutaneous leishmaniasis of the guinea pig, Leishmania enriettii n. sp. Hospital (Rio J). 1948;33(1):7–25 (in Portuguese).
Jariyapan N, Daroontum T, Jaiwong K, Chanmol W, Intakhan N, Sor-Suwan S, Siriyasatien P, Somboon P, Bates MD, Bates PA. Leishmania (Mundinia) orientalis n. sp. (Trypanosomatidae), a parasite from Thailand responsible for localised cutaneous leishmaniasis. Parasit Vectors. 2018;11(1):351.
Barratt J, Kaufer A, Peters B, Craig D, Lawrence A, Roberts T, Lee R, McAuliffe G, Stark D, Ellis J. Isolation of novel trypanosomatid, Zelonia australiensis sp. nov. (Kinetoplastida: Trypanosomatidae) provides support for a Gondwanan origin of dixenous parasitism in the Leishmaniinae. PLOS Negl Trop Dis. 2017;11(1):e0005215.
Desbois N, Pratlong F, Quist D, Dedet JP. Leishmania (Leishmania) martiniquensis n. sp. (Kinetoplastida: Trypanosomatidae), description of the parasite responsible for cutaneous leishmaniasis in Martinique Island (French West Indies). Parasite. 2014;21:12.
Kwakye-Nuako G, Mosore MT, Duplessis C, Bates MD, Puplampu N, Mensah-Attipoe I, Desewu K, Afegbe G, Asmah RH, Jamjoom MB, et al. First isolation of a new species of Leishmania responsible for human cutaneous leishmaniasis in Ghana and classification in the Leishmania enriettii complex. Int J Parasitol. 2015;45(11):679–84.
Machado MI, Milder RV, Pacheco RS, Silva M, Braga RR, Lainson R. Naturally acquired infections with Leishmania enriettii Muniz and Medina 1948 in Guinea-pigs from São Paulo, Brazil. Parasitology. 1994;109:135–8.
Paranaiba LF, Pinheiro LJ, Torrecilhas AC, Macedo DH, Menezes-Neto A, Tafuri WL, Soares RP. Leishmania enriettii (Muniz & Medina, 1948): a highly diverse parasite is here to stay. PLoS Pathog. 2017;13(5):e1006303.
Dougall A, Shilton C, Low Choy J, Alexander B, Walton S. New reports of Australian cutaneous leishmaniasis in northern Australian macropods. Epidemiol Infect. 2009;137(10):1516–20.
Rose K, Curtis J, Baldwin T, Mathis A, Kumar B, Sakthianandeswaren A, Spurck T, Low Choy J, Handman E. Cutaneous leishmaniasis in red kangaroos: isolation and characterisation of the causative organisms. Int J Parasitol. 2004;34(6):655–64.
Lobsiger L, Muller N, Schweizer T, Frey CF, Wiederkehr D, Zumkehr B, Gottstein B. An autochthonous case of cutaneous bovine leishmaniasis in Switzerland. Vet Parasitol. 2010;169(3–4):408–14.
Reuss SM, Dunbar MD, Calderwood Mays MB, Owen JL, Mallicote MF, Archer LL, Wellehan JF Jr. Autochthonous Leishmania siamensis in horse, Florida, USA. Emerg Infect Dis. 2012;18(9):1545–7.
Müller N, Welle M, Lobsiger L, Stoffel MH, Boghenbor KK, Hilbe M, Gottstein B, Frey CF, Geyer C, von Bomhard W. Occurrence of Leishmania sp. in cutaneous lesions of horses in Central Europe. Vet Parasitol. 2009;166(3–4):346–51.
Bualert L, Charungkiattikul W, Thongsuksai P, Mungthin M, Siripattanapipong S, Khositnithikul R, Naaglor T, Ravel C, El Baidouri F, Leelayoova S. Autochthonous disseminated dermal and visceral leishmaniasis in an AIDS patient, southern Thailand, caused by Leishmania siamensis. Am J Trop Med Hyg. 2012;86(5):821–4.
Dedet JP, Roche B, Pratlong F, Cales-Quist D, Jouannelle J, Benichou JC, Huerre M. Diffuse cutaneous infection caused by a presumed monoxenous trypanosomatid in a patient infected with HIV. Trans R Soc Trop Med Hyg. 1995;89(6):644–6.
Chicharro C, Alvar J. Lower trypanosomatids in HIV/AIDS patients. Ann Trop Med Parasitol. 2003;97(Suppl 1):75–8.
Dedet JP, Pratlong F. Leishmania, Trypanosoma and monoxenous trypanosomatids as emerging opportunistic agents. J Eukaryot Microbiol. 2000;47(1):37–9.
Kraeva N, Butenko A, Hlaváčová J, Kostygov A, Myškova J, Grybchuk D, Leštinová T, Votýpka J, Volf P, Opperdoes F, et al. Leptomonas seymouri: adaptations to the dixenous life cycle analyzed by genome sequencing, transcriptome profiling and co-infection with Leishmania donovani. PLOS Pathog. 2015;11(8):e1005127.
Ghosh S, Banerjee P, Sarkar A, Datta S, Chatterjee M. Coinfection of Leptomonas seymouri and Leishmania donovani in Indian leishmaniasis. J Clin Microbiol. 2012;50(8):2774–8.
Kostygov AY, Butenko A, Yurchenko V. On monoxenous trypanosomatids from lesions of immunocompetent patients with suspected cutaneous leishmaniasis in Iran. Tropical Med Int Health. 2019;24(1):127–8.
Dougall AM, Alexander B, Holt DC, Harris T, Sultan AH, Bates PA, Rose K, Walton SF. Evidence incriminating midges (Diptera: Ceratopogonidae) as potential vectors of Leishmania in Australia. Int J Parasitol. 2011;41(5):571–9.
Harkins KM, Schwartz RS, Cartwright RA, Stone AC. Phylogenomic reconstruction supports supercontinent origins for Leishmania. Infect Genet Evol. 2016;38:101–9.
Coughlan S, Mulhair P, Sanders M, Schonian G, Cotton JA, Downing T. The genome of Leishmania adleri from a mammalian host highlights chromosome fission in Sauroleishmania. Sci Rep. 2017;7:43747.
Raymond F, Boisvert S, Roy G, Ritt JF, Legare D, Isnard A, Stanke M, Olivier M, Tremblay MJ, Papadopoulou B, et al. Genome sequencing of the lizard parasite Leishmania tarentolae reveals loss of genes associated to the intracellular stage of human pathogenic species. Nucleic Acids Res. 2012;40(3):1131–47.
Coughlan S, Taylor AS, Feane E, Sanders M, Schonian G, Cotton JA, Downing T. Leishmania naiffi and Leishmania guyanensis reference genomes highlight genome structure and gene evolution in the Viannia subgenus. R Soc Open Sci. 2018;5(4):172212.
Llanes A, Restrepo CM, Del Vecchio G, Anguizola FJ, Lleonart R. The genome of Leishmania panamensis: insights into genomics of the L. (Viannia) subgenus. Sci Rep. 2015;5:8550.
Rogers MB, Hilley JD, Dickens NJ, Wilkes J, Bates PA, Depledge DP, Harris D, Her Y, Herzyk P, Imamura H, et al. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 2011;21(12):2129–42.
Valdivia HO, Reis-Cunha JL, Rodrigues-Luiz GF, Baptista RP, Baldeviano GC, Gerbasi RV, Dobson DE, Pratlong F, Bastien P, Lescano AG, et al. Comparative genomic analysis of Leishmania (Viannia) peruviana and Leishmania (Viannia) braziliensis. BMC Genomics. 2015;16:715.
Peacock CS, Seeger K, Harris D, Murphy L, Ruiz JC, Quail MA, Peters N, Adlem E, Tivey A, Aslett M, et al. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat Genet. 2007;39(7):839–47.
Cantacessi C, Dantas-Torres F, Nolan MJ, Otranto D. The past, present, and future of Leishmania genomics and transcriptomics. Trends Parasitol. 2015;31(3):100–8.
Maslov DA, Lukeš J, Jirků M, Simpson L. Phylogeny of trypanosomes as inferred from the small and large subunit rRNAs: implications for the evolution of parasitism in the trypanosomatid protozoa. Mol Biochem Parasitol. 1996;75(2):197–205.
Kostygov AY, Grybchuk-Ieremenko A, Malysheva MN, Frolov AO, Yurchenko V. Molecular revision of the genus Wallaceina. Protist. 2014;165(5):594–604.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10.
Ishemgulova A, Hlavacova J, Majerova K, Butenko A, Lukes J, Votypka J, Volf P, Yurchenko V. CRISPR/Cas9 in Leishmania mexicana: a case study of LmxBTN1. PLoS One. 2018;13(2):e0192723.
Flegontov P, Butenko A, Firsov S, Kraeva N, Eliáš M, Field MC, Filatov D, Flegontova O, Gerasimov ES, Hlaváčová J, et al. Genome of Leptomonas pyrrhocoris: a high-quality reference for monoxenous trypanosomatids and new insights into evolution of Leishmania. Sci Rep. 2016;6:23704.
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–2.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–77.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52.
Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9(4):357–9.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc. 2012;7(3):562–78.
Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29(8):1072–5.
Steinbiss S, Silva-Franco F, Brunk B, Foth B, Hertz-Fowler C, Berriman M, Otto TD. Companion: a web server for annotation and analysis of parasite genomes. Nucleic Acids Res. 2016;44(W1):W29–34.
Soderlund C, Nelson W, Shoemaker A, Paterson A. SyMAP: a system for discovering and viewing syntenic regions of FPC maps. Genome Res. 2006;16(9):1159–68.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–2.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
Rimmer A, Phan H, Mathieson I, Iqbal Z, Twigg SR, Wilkie AO, McVean G, Lunter G. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet. 2014;46(8):912–8.
Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16:157.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7.
Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7(10):e1002195.
Capella-Gutiérrez S, Silla-Martinez JM, Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25(15):1972–3.
Minh BQ, Nguyen MA, von Haeseler A. Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol. 2013;30(5):1188–95.
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32(1):268–74.
Lartillot N, Rodrigue N, Stubbs D, Richer J. PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst Biol. 2013;62(4):611–5.
Csuros M. Count: evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics. 2010;26(15):1910–2.
Conesa A, Götz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–6.
Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol. 2016;428(4):726–31.
Lex A, Gehlenborg N, Strobelt H, Vuillemot R, Pfister H. UpSet: visualization of intersecting sets. IEEE Trans Vis Comput Graph. 2014;20(12):1983–92.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger satasets. Mol Biol Evol. 2016;33(7):1870–4.
Butenko A, Vieira TDS, Frolov AO, Opperdoes FR, Soares RP, Kostygov AY, Lukeš J, Yurchenko V. Leptomonas pyrrhocoris: genomic insight into parasite's physiology. Curr Genomics. 2018;19(2):150–6.
Aslett M, Aurrecoechea C, Berriman M, Brestelli J, Brunk BP, Carrington M, Depledge DP, Fischer S, Gajria B, Gao X, et al. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 2010;38(Database issue):D457–62.
Kangussu-Marcolino MM, de Paiva RM, Araujo PR, de Mendonca-Neto RP, Lemos L, Bartholomeu DC, Mortara RA, daRocha WD, Teixeira SM. Distinct genomic organization, mRNA expression and cellular localization of members of two amastin sub-families present in Trypanosoma cruzi. BMC Microbiol. 2013;13:10.
de Paiva RM, Grazielle-Silva V, Cardoso MS, Nakagaki BN, Mendonca-Neto RP, Canavaci AM, Souza Melo N, Martinelli PM, Fernandes AP, daRocha WD, et al. Amastin knockdown in Leishmania braziliensis affects parasite-macrophage interaction and results in impaired viability of intracellular amastigotes. PLoS Pathog. 2015;11(12):e1005296.
Lypaczewski P, Hoshizaki J, Zhang WW, McCall LI, Torcivia-Rodriguez J, Simonyan V, Kaur A, Dewar K, Matlashewski G. A complete Leishmania donovani reference genome identifies novel genetic variations associated with virulence. Sci Rep. 2018;8(1):16549.
Jackson AP. The evolution of amastin surface glycoproteins in trypanosomatid parasites. Mol Biol Evol. 2010;27(1):33–45.
Pech-Canul AC, Monteon V, Solis-Oviedo RL. A brief view of the surface membrane proteins from Trypanosoma cruzi. J Parasitol Res. 2017;2017:3751403.
Ilg T, Handman E, Stierhof YD. Proteophosphoglycans from Leishmania promastigotes and amastigotes. Biochem Soc Trans. 1999;27(4):518–25.
Hong Y, Kinoshita T. Trypanosome glycosylphosphatidylinositol biosynthesis. Korean J Parasitol. 2009;47(3):197–204.
Sacks DL, Saraiva EM, Rowton E, Turco SJ, Pimenta PF. The role of the lipophosphoglycan of Leishmania in vector competence. Parasitology. 1994;108(Suppl):S55–62.
McConville MJ, Turco SJ, Ferguson MA, Sacks DL. Developmental modification of lipophosphoglycan during the differentiation of Leishmania major promastigotes to an infectious stage. EMBO J. 1992;11(10):3593–600.
Dobson DE, Scholtes LD, Valdez KE, Sullivan DR, Mengeling BJ, Cilmi S, Turco SJ, Beverley SM. Functional identification of galactosyltransferases (SCGs) required for species-specific modifications of the lipophosphoglycan adhesin controlling Leishmania major-sand fly interactions. J Biol Chem. 2003;278(18):15523–31.
Dobson DE, Scholtes LD, Myler PJ, Turco SJ, Beverley SM. Genomic organization and expression of the expanded SCG/L/R gene family of Leishmania major: internal clusters and telomeric localization of SCGs mediating species-specific LPG modifications. Mol Biochem Parasitol. 2006;146(2):231–41.
Sacks DL, Modi G, Rowton E, Spath G, Epstein L, Turco SJ, Beverley SM. The role of phosphoglycans in Leishmania-sand fly interactions. Proc Natl Acad Sci U S A. 2000;97(1):406–11.
Soares RP, Cardoso TL, Barron T, Araujo MS, Pimenta PF, Turco SJ. Leishmania braziliensis: a novel mechanism in the lipophosphoglycan regulation during metacyclogenesis. Int J Parasitol. 2005;35(3):245–53.
de Assis RR, Ibraim IC, Nogueira PM, Soares RP, Turco SJ. Glycoconjugates in New World species of Leishmania: polymorphisms in lipophosphoglycan and glycoinositolphospholipids and interaction with hosts. Biochim Biophys Acta. 2012;1820(9):1354–65.
Vickers TJ, Orsomando G, de la Garza RD, Scott DA, Kang SO, Hanson AD, Beverley SM. Biochemical and genetic analysis of methylenetetrahydrofolate reductase in Leishmania metabolism and virulence. J Biol Chem. 2006;281(50):38150–8.
Vickers TJ, Beverley SM. Folate metabolic pathways in Leishmania. Essays Biochem. 2011;51:63–80.
Opperdoes FR, Coombs GH. Metabolism of Leishmania: proven and predicted. Trends Parasitol. 2007;23(4):149–58.
Chang KP, Chang CS, Sassa S. Heme biosynthesis in bacterium-protozoon symbioses: enzymic defects in host hemoflagellates and complemental role of their intracellular symbiotes. Proc Natl Acad Sci U S A. 1975;72(8):2979–83.
Opperdoes FR, Butenko A, Flegontov P, Yurchenko V, Lukeš J. Comparative metabolism of free-living Bodo saltans and parasitic trypanosomatids. J Eukaryot Microbiol. 2016;63(5):657–78.
Kořený L, Sobotka R, Kovářová J, Gnipová A, Flegontov P, Horváth A, Oborník M, Ayala FJ, Lukeš J. Aerobic kinetoplastid flagellate Phytomonas does not require heme for viability. Proc Natl Acad Sci U S A. 2012;109(10):3808–13.
Cenci U, Moog D, Curtis BA, Tanifuji G, Eme L, Lukes J, Archibald JM. Heme pathway evolution in kinetoplastid protists. BMC Evol Biol. 2016;16(1):109.
Kořený L, Oborník M, Lukeš J. Make it, take it, or leave it: heme metabolism of parasites. PLoS Pathog. 2013;9(1):e1003088.
Silva FM, Kostygov AY, Spodareva VV, Butenko A, Tossou R, Lukes J, Yurchenko V, Alves JMP. The reduced genome of Candidatus Kinetoplastibacterium sorsogonicusi, the endosymbiont of Kentomonas sorsogonicus (Trypanosomatidae): loss of the haem-synthesis pathway. Parasitology. 2018. https://doi.org/10.1017/S003118201800046X.
Kim EJ, Oh EK, Lee JK. Role of HemF and HemN in the heme biosynthesis of Vibrio vulnificus under S-adenosylmethionine-limiting conditions. Mol Microbiol. 2015;96(3):497–512.
Sen Santara S, Roy J, Mukherjee S, Bose M, Saha R, Adak S. Globin-coupled heme containing oxygen sensor soluble adenylate cyclase in Leishmania prevents cell death during hypoxia. Proc Natl Acad Sci U S A. 2013;110(42):16790–5.
Kutateladze TG. Phosphatidylinositol 3-phosphate recognition and membrane docking by the FYVE domain. Biochim Biophys Acta. 2006;1761(8):868–77.
Kutateladze TG, Ogburn KD, Watson WT, de Beer T, Emr SD, Burd CG, Overduin M. Phosphatidylinositol 3-phosphate recognition by the FYVE domain. Mol Cell. 1999;3(6):805–11.
Wilson V, Southgate B. Lizard Leishmania. In: Lumsden W, Evans DA, editors. Biology of Kinetoplastida. New York: Academic; 1979. p. 242–68.
Acknowledgements
We are grateful to the members of our laboratories for stimulating discussions.
Funding
Support from the ERD Funds (project OPVVV 16_019/0000759 to VY, PV, JS, AYK, and JL), University of Ostrava (project SGS08/PrF/2019 to LP, DHM, and DŽ), Russian Science Foundation (project 19–15- 00054 to VY), and Grant Agency of Czech Republic (projects 17-10656S to VY) is kindly acknowledged. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
AB, PV and VY conceived the study. AB, AYK, JS, YK, TB, LP, DHM, DŽ, JL, PAB, PV, FRO, VY analyzed and interpreted the whole-genome data. AB and VY were major contributors in writing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
VY is an Associate Editor of BMC Genomics. Other authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
BlobTools statistics for L. (M.) enriettii MCAV/BR/1945/LV90 before filtering.
Additional file 2: Figure S2.
BlobTools statistics for L. (M.) enriettii MCAV/BR/1945/LV90 after filtering.
Additional file 3: Figure S3.
BlobTools statistics for L. (M.) macropodum MMAC/AU/2004/AM-2004 before filtering.
Additional file 4: Figure S4.
BlobTools statistics for L. (M.) macropodum MMAC/AU/2004/AM-2004 after filtering.
Additional file 5: Figure S5.
BlobTools statistics for L. (M.) martiniquensis MHOM/MQ/1992/MAR1 before filtering.
Additional file 6: Figure S6.
BlobTools statistics for L. (M.) martiniquensis MHOM/MQ/1992/MAR1 after filtering.
Additional file 7: Figure S7.
CovPlot statistics for the final assembly of L. (M.) enriettii MCAV/BR/1945/LV90.
Additional file 8: Figure S8.
CovPlot statistics for the final assembly of L. (M.) macropodum MMAC/AU/2004/AM-2004.
Additional file 9: Figure S9.
CovPlot statistics for the final assembly of L. (M.) martiniquensis MHOM/MQ/1992/MAR1.
Additional file 10: Figure S10.
Plot showing the distribution of genomic read coverage values for the genome assemblies of L. (M.) enriettii (blue line), L. (M.) martiniquensis (orange), L. (M.) macropodum (green).
Additional file 11: Figure S11.
Panel (A). Schematic representation of the two-way synteny between the genomes of Leishmania (Mundinia) strains sequenced in this study and the ones available in TriTrypDB, as well as between Leishmania (Mundinia) and L. major Friedlin. Corresponding syntenic blocks are connected with red ribbons. In each case scaffolds of two compared strains/species are filled with different colors and are separated by a blank space. Only the chromosomes which actually have synteny blocks are shown. Panel (B). Summary statistics for pairwise synteny analyses among Leishmania (Mundinia) strains and the reference genome sequence of L. major Friedlin.
Additional file 12: Figure S12.
Maximum-Likelihood phylogenetic tree of trypanosomatid amastins. The tree was inferred using IQ-TREE v.1.5.3 with the JTT + I + G4 model and 1000 bootstrap replicates. The support values are in the following format: SH-aLRT support (%)/bootstrap support (%).
Additional file 13: Figure S13.
Maximum-Likelihood phylogenetic tree of trypanosomatid phosphatydylinositol glycan class Y (PIG-Y) sequences. The tree was inferred using IQ-TREE v.1.5.3 with the JTT + I + G4 model and 1000 bootstrap replicates. The support values are in the following format: SH-aLRT support (%)/bootstrap support (%).
Additional file 14: Figure S14.
Maximum-Likelihood phylogenetic tree of trypanosomatid side chain galactosyltransferases (SCGs) and side chain arabinosyltransferases (SCAs) sequences. The tree was inferred using IQ-TREE v.1.5.3 with 1000 bootstrap replicates and VT + F + I + G4 and JTT + F + G4 models for SCGs and SCAs, respectively. The support values are in the following format: SH-aLRT support (%)/bootstrap support (%). Reference SCGs and SCAs of L. major are highlighted in color.
Additional file 15: Figure S15.
Maximum-Likelihood phylogenetic tree of ferrochelatase sequences. The tree was constructed using IQ-TREE v.1.5.3 with 1000 bootstrap replicates and JTT + I + G4 model. The support values are in the following format: SH-aLRT support (%)/bootstrap support (%).
Additional file 16: Table S1.
Dataset used in this study. Organisms, whose genomes and transcriptomes were sequenced in this work, are in bold.
Additional file 17: Table S2.
Estimates of evolutionary divergence among putative amastin sequences. The number of amino acid differences per site from between sequences are shown. The analysis involved 450 amino acid sequences. All ambiguous positions were removed for each sequence pair. There were a total of 1532 positions in the final dataset. Evolutionary analyses were conducted in MEGA7.
Additional file 18: Table S3.
Estimates of evolutionary divergence among putative side chain galactosyltransferase sequences. The number of amino acid differences per site from between sequences are shown. The analysis involved 87 amino acid sequences. All positions with less than 80% site coverage were eliminated. Less than 20% alignment gaps, missing data and ambiguous bases were allowed at any position. There were a total of 476 positions in the final dataset. Evolutionary analyses were conducted in MEGA7. The presence of n/c in the results denotes cases in which it was not possible to estimate evolutionary distances.
Additional file 19: Table S4.
Estimates of evolutionary divergence among putative side chain arabinosyltransferase sequences. The number of amino acid differences per site from between sequences are shown. The analysis involved 20 amino acid sequences. All positions with less than 80% site coverage were eliminated. Less than 20% alignment gaps, missing data and ambiguous bases were allowed at any position. There were total of 694 positions in the final dataset. Evolutionary analyses were conducted in MEGA7.
Additional file 20: Table S5.
Estimates of evolutionary divergence among putative phosphatydylinositol glycan class Y (PIG-Y) sequences. The number of amino acid differences per site from between sequences are shown. The analysis involved 14 amino acid sequences. All ambiguous positions were removed for each sequence pair. There were a total of 128 positions in the final dataset. Evolutionary analyses were conducted in MEGA7.
Additional file 21: Table S6.
Estimates of evolutionary divergence among putative ferrochelatase sequences. The number of amino acid differences per site from between sequences are shown. The analysis involved 45 amino acid sequences. All positions with less than 80% site coverage were eliminated. Less than 20% alignment gaps, missing data and ambiguous bases were allowed at any position. There were a total of 247 positions in the final dataset. Evolutionary analyses were conducted in MEGA7. The presence of n/c in the results denotes cases in which it was not possible to estimate evolutionary distances.
Additional file 22: Table S7.
Summary statistics for Mundinia genomes sequenced in this study and several publicly available trypanosomatid genome assemblies.
Additional file 23: Table S8.
Ploidy estimates for fifty longest scaffolds and all pseudochromosome level sequences of Leishmania (Mundinia) spp. sequenced in this study.
Additional file 24: Table S9.
Gene gains and losses at the Leishmania (Mundinia) node.
Additional file 25: Table S10.
Gene gains and losses at the Leishmania/ Sauroleishmania/ Viannia node.
Additional file 26: Table S11.
Gene family expansion and contractions at the Leishmania (Mundinia) node.
Additional file 27: Table S12.
Gene family expansion and contractions at the Leishmania/ Sauroleishmania/ Viannia node.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Butenko, A., Kostygov, A.Y., Sádlová, J. et al. Comparative genomics of Leishmania (Mundinia). BMC Genomics 20, 726 (2019). https://doi.org/10.1186/s12864-019-6126-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-6126-y