
Abstract
Background
Cytoplasmic incompatibility (CI) is the most common phenotype induced by endosymbiont Wolbachia and results in embryonic lethality when Wolbachia-modified sperm fertilize eggs without Wolbachia. However, eggs carrying the same strain of Wolbachia can rescue this embryonic death, thus producing viable Wolbachia-infected offspring. Hence Wolbachia can be transmitted mainly by hosts’ eggs. One of the models explaining CI is “titration-restitution”, which hypothesized that Wolbachia titrated-out some factors from the sperm and the Wolbachia in the egg would restitute the factors after fertilization. However, how infected eggs rescue CI and how hosts’ eggs ensure the proliferation and transmission of Wolbachia are not well understood.
Results
By RNA-seq analyses, we first compared the transcription profiles of Drosophila melanogaster adult ovaries with and without the wMel Wolbachia and identified 149 differentially expressed genes (DEGs), of which 116 genes were upregulated and 33 were downregulated by Wolbachia infection. To confirm the results obtained from RNA-seq and to screen genes potentially associated with reproduction, 15 DEGs were selected for quantitative RT-PCR (qRT-PCR). Thirteen genes showed the same changing trend as RNA-seq analyses. To test whether these genes are associated with CI, we also detected their expression levels in testes. Nine of them exhibited different changing trends in testes from those in ovaries. To investigate how these DEGs were regulated, sRNA sequencing was performed and identified seven microRNAs (miRNAs) that were all upregulated in fly ovaries by Wolbachia infection. Matching of miRNA and mRNA data showed that these seven miRNAs regulated 15 DEGs. Wolbachia-responsive genes in fly ovaries were involved in biological processes including metabolism, transportation, oxidation-reduction, immunity, and development.
Conclusions
Comparisons of mRNA and miRNA data from fly ovaries revealed 149 mRNAs and seven miRNAs that exhibit significant changes in expression due to Wolbachia infection. Notably, most of the DEGs showed variation in opposite directions in ovaries versus testes in the presence of Wolbachia, which generally supports the “titration-restitution” model for CI. Furthermore, genes related to metabolism were upregulated, which may benefit maximum proliferation and transmission of Wolbachia. This provides new insights into the molecular mechanisms of Wolbachia-induced CI and Wolbachia dependence on host ovaries.
Similar content being viewed by others

Background
Wolbachia are widespread endosymbionts that frequently infect many insect species. They are well-known for their ability to manipulate host reproduction for their own propagation [1, 2]. Sperm-egg cytoplasmic incompatibility (CI) is the most common reproductive phenotype induced by Wolbachia, and it results in sterility or notably low hatch rates when mating occurs between Wolbachia-infected males and uninfected females. This indicates that infected males are “dead-end” hosts for Wolbachia; however, females carrying the same strain of Wolbachia can rescue zygotic lethality associated with CI [1, 3]. Thus, Wolbachia-mediated CI can be used as promising tools for the control of pests and disease vectors. Although this phenomenon has been known for around 45 years [4], the underlying molecular mechanisms of CI and how CI can be rescued are still not yet well understood and pose a great challenge to researchers who are working on it.
Cytological studies have shown that in CI embryos, paternal chromatin condensation and segregation defects during the first mitotic division following fertilization are the main causes of early embryonic mortality [5, 6]. Therefore, sperms from Wolbachia-infected males may be modified during spermatogenesis. Bourtzis et al. proposed a “modification/rescue” model to explain the production and rescue of CI, in which Wolbachia can modify male sperm during spermatogenesis, and females carrying the same Wolbachia can rescue such modification after insemination [7]. Thereafter several models were proposed to translate the “modification/rescue” concept [8, 9]. One of them is “titration-restitution” model, which proposed that during spermatogenesis, Wolbachia could titrate some essential components from the sperm, and the same strain of Wolbachia in the egg may compensate these critical factors, allowing embryogenesis to proceed normally [8]. According to this model, “modification” and “rescue” can be determined by the same gene(s) or by different genes. In the former case, the transition from titration to restitution after fertilization will be triggered by infected female hosts. In the latter case, one gene codes factors for titration and the other codes factors for restitution [8]. Some evidences support the former case. For instance, Ance, coding for angiotensin-converting enzyme, has been shown to be significantly upregulated in infected females but notably downregulated in infected males of Drosophila simulans [10]. Our previous work revealed that the expression level of Hira, coding for a negative regulator of histone gene expression, was higher in females, but lower in males when compared to uninfected D. melanogaster [11]. Recently, several studies have demonstrated that Wolbachia can secret gene products to host cells that may directly manipulate host reproduction [12,13,14,15,16]. For example, two adjacent genes, cifA and cifB, were identified from the wMel strain to be responsible for the wMel-induced sperm modification of CI, and cifA expression in ovaries can rescue CI in D. melanogaster [14, 16, 17]. Thus, Shropshire et al. established a “Two-by-One” model [17], which could partially support the latter case that shift from titration to restitution could be determined by different genes.
As Wolbachia-associated embryonic lethality is limited to paternal chromosomes, studies related to CI have mainly focused on males [11, 18,19,20,21]. For example, we previously found that the decreased Hira expression in male D. melanogaster was closely related to Wolbachia-induced CI [11]. Microarray analyses of the 3rd instar larval testes of D. melanogaster with and without Wolbachia revealed that among those DEGs, most genes putatively involved in reproduction exhibited downregulation in the presence of Wolbachia [18]. Comparative proteomics of the spermatheca and seminal receptacle (containing sperm proteins and seminal fluid proteins from their mates) from uninfected females mated with Wolbachia-infected or uninfected males identified many proteins including seminal fluid proteins that were downregulated due to Wolbachia infection [20]. Landmann et al. found that in CI embryos, a delay of histone H3.3 and H4 deposition occurred on the sperm nuclei during male pronuclei formation, which may cause the asynchronous development of male and female pronuclei and thus embryonic death [22]. However, there are few studies on Wolbachia-infected ovaries [23]. How Wolbachia-infected eggs can rescue CI, and how Wolbachia modify Drosophila ovary to benefit for their own survival and propagation are unclear.
Considering that Wolbachia-infected eggs can rescue embryonic lethality when fertilized with sperm produced by Wolbachia-infected males, but Wolbachia-uninfected eggs cannot, and that Wolbachia can be transferred to progeny mainly by the egg, we applied RNA-seq technology to compare the transcriptional profiles as well as sRNAs expression patterns between Wolbachia-infected and uninfected adult ovaries of D. melanogaster. We identified 149 genes with at least a 2-fold change in expression (p < 5%). We also identified seven significantly differentially expressed miRNAs, which were all significantly upregulated in ovaries harboring Wolbachia. The target genes of differentially expressed miRNAs were further identified. The changed expression patterns of some genes caused by Wolbachia infection in ovaries and testes were compared. In general, our results, together with previous data, support the titration-restitution hypothesis for CI. These findings may provide new insights into Wolbachia/host interactions and especially the molecular mechanisms of CI induced by Wolbachia.
Results
Identification of mRNAs involved in Wolbachia infection in Drosophila ovaries
To investigate the rescue mechanisms involved in CI induced by Wolbachia and the impact of Wolbachia on maternal transmission, we first compared the mRNA profiles between Wolbachia-infected and uninfected Drosophila ovaries by RNA-seq. A total of 60,847,382 sequence reads for Dmel T (Drosophila melanogaster without Wolbachia) ovaries and 54,620,985 for Dmel wMel (D. melanogaster infected wMel Wolbachia) ovaries were generated. An overview of sequencing and assembly is outlined in Table 1. After removal of adaptor sequences, ambiguous reads and low-quality reads, 58,795,543 high-quality clean reads (96.6% of the raw data) of Dmel T remained, while 53,645,601 high-quality clean reads (98.2% of the raw data) of Dmel wMel remained. Eighty-eight percent of clean reads had Phred-like quality scores at the Q30 level (an error probability of 0.001).
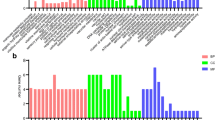
A total of 149 differentially expressed genes (DEGs) were identified, 116 of which were upregulated and 33 of which were downregulated in the presence of Wolbachia (Fig. 1a, Additional file 1). When these DEGs were subjected to KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis, we found that starch and sucrose metabolic pathways had the largest number of DEGs. Furthermore, the TGF-β signaling pathway, galactose metabolism, Wnt signaling pathway, ubiquitin-mediated proteolysis, and protein processing in endoplasmic reticulum were also enriched (Fig. 1b, Additional file 2). Gene Ontology (GO) annotation enrichment revealed that among the DEGs with known biological process, metabolism, organism process, cellular process, localization, transportation, and development were most prevalent (Additional file 3).

Volcanoplot (a) and KEGG pathway (b) of differentially expressed genes in the Drosophila melanogaster ovary due to the infection of Wolbachia. (a) Distribution of the relative expression of genes in the infected versus uninfected fly ovaries. Green dots represent downregulated genes; red dots represent upregulated genes and blue dots represent unchanged genes. (b) The 20 most enriched KEGG pathways based on the differentially expressed genes in the presence of Wolbachia. The x-axis shows the rich factor; the y-axis shows the pathway names. The size of each point represents the number of genes enriched in a particular pathway. The larger the rich factor and the smaller the q-value, the more significant the degree of enrichment. Dmel T: Drosophila melanogaster treated with tetracycline (without Wolbachia); Dmel wMel: Drosophila melanogaster infected with wMel Wolbachia
It is interesting that some genes encoding partners of cytochrome P450 (CYP) showed differential expression patterns. Four genes, including Cyp4p1, Cyp12a5, Cyp6g1, and Cyp12a4, were identified to be upregulated, while Cyp6a13 was downregulated in the presence of Wolbachia (Additional file 1). These five genes belong to the CYP, E-class, group I according to FlyBase and are involved in oxidation-reduction processes (http://flybase.bio.indiana.edu).
qRT-PCR validation of some DEGs induced by Wolbachia infection in ovaries and testes
In order to confirm the results observed during RNA-seq analysis, 15 differentially expressed genes based on large differences in expression in the RNA-seq analyses in addition to genes potentially associated with reproduction were selected for qRT-PCR to further investigate their expression profiles (Table 2). The results demonstrated that most of the genes (13) whose expression in ovaries measured by qRT-PCR exhibited similar changes as in the RNA-seq, with 6 genes (RpL22-like, uif, CG32054, dany, otk2, and CG10659) upregulated and 7 genes (AttC, CG6435, Twdlβ, pgant8, CG5111, Def, and CG18258) downregulated (Fig. 2a).

qRT-PCR validation of differentially expressed genes identified by RNA sequencing. a: gene expression in ovaries; b: gene expression in testes. A two-tailed Student’s t-test was performed to compare the significance between Wolbachia-infected (Dmel wMel) and uninfected (Dmel T) fly ovaries (a) and testes (b) of candidate genes. Red color indicates upregulation; Blue color indicates downregulation; Black color indicates not significantly different.“/” represents the relative value; bars indicate standard error; “*” and “**” indicate significant differences with p < 0.05 and p < 0.01, respectively. df = 4, n = 3
Considering that young Wolbachia-infected male flies can induce high level of CI [11, 24], to test whether the gene expression levels change due to Wolbachia infection is in a reverse trend in testes, we also detected the expression level of these genes in 1-day-old fly testes. Four genes (dany, AttC, pgant8, and Def) were upregulated and four genes (RpL22-like, CG6435, CG5111, and CG18258) were downregulated in the presence of Wolbachia (Fig. 2b). Of these selected genes, RpL22-like showed the highest upregulation in Wolbachia-infected ovaries (around 10 times) and downregulation in Wolbachia-infected testes, which were similar to the expression patterns of Ance and Hira [10, 11]. Other genes of interest including AttC, pgant8, and Def were downregulated in Wolbachia-infected ovaries but upregulated in infected testes.
sRNA annotations in Dmel wMel and Dmel T ovaries
An increasing body of evidence suggests that non-coding RNAs, including small RNAs (sRNAs), play a critical role in regulating gene expression and thus in reproduction and development [25, 26]. To study the responses of sRNAs in ovaries to Wolbachia infection, two sRNA libraries were constructed from Dmel T and Dmel wMel virgin fly ovaries with an average of 8.11 million raw clean reads, ranging from 18 nt to 35 nt in length. The peak size was 30 nt, followed by 29 nt and 31 nt (Fig. 3), indicating that PIWI-interacting RNAs (piRNAs) might be the major component of ovary sRNAs. An overview of sequencing and assembly is outlined in Table 3. A total of 7,700,845 high-quality clean reads of Dmel T remained; 7,690,372 high-quality clean reads of Dmel wMel remained. Nighty-6 % of clean read data had Phred-like quality scores at the Q30 level. The sRNAs were divided into twelve classes, such as miRNAs, piRNAs and rRNAs (Fig. 4). The percentage of piRNAs in unique reads in Dmel T and Dmel wMel were highest: 14.12% (including 11.54% of novel piRNAs) and 13.29% (including 10.77% of novel piRNAs), respectively. This is consistent with Fig. 3 where piRNAs were the major component of sRNAs in the ovary. Furthermore, the percentage of known miRNAs in unique reads in Dmel T and Dmel wMel were 0.61 and 0.65%, and rRNA accounted for 6.65 and 9.30% in Dmel T and Dmel wMel, respectively (Fig. 4a and b).

The length of distribution of small RNAs (sRNAs) in Dmel T and Dmel wMel ovaries. Dmel T: Drosophila melanogaster treated with tetracycline (without Wolbachia); Dmel wMel: D. melanogaster infected with wMel Wolbachia. nt: nucleotides

Classification of sRNAs in Dmel T and Dmel wMel ovaries. Dmel T: Drosophila melanogaster treated with tetracycline (without Wolbachia); Dmel wMel: D. melanogaster infected with wMel Wolbachia. miRNA: microRNA; piRNA: Piwi-interacting RNA; snRNA: small nuclear RNA; sonRNA: small nucleolar RNA; rRNA: ribosome RNA; tRNA: transfer RNA; repeat: repeative sRNA sequences; exon:+: exon sense; exon:-: exon antisense; intron:+: intron sense; intron:-: intron antisense; other: unannotated sRNA
Identification of differentially expressed miRNAs involved in Drosophila ovaries and testes
In Wolbachia-infected female ovaries, seven miRNAs were identified as upregulated compared with uninfected controls (Table 4). However, no significantly downregulated miRNAs were observed. To verify the results of sRNA-seq, the expression profiles of these seven miRNAs were further measured by qRT-PCR. Five miRNAs (dme-miR-982-5p, dme-miR-983-5p, dme-miR-982-3p, miR-984-5p, and dme-miR-318-3p) exhibited significantly upregulated in the presence of Wolbachia (Fig. 5), which is in conformity with sRNA-seq analyses. However, dme-miR-956-3p showed no significant change. We could not detect dme-miR-983-3p expression in ovaries by qRT-PCR. As for the testes, dme-miR-983-5p manifested notable upregulation, whereas dme-miR-318-3p and dme-miR-956-3p showed significant downregulation due to Wolbachia infection (Fig. 5b, e, f). However, dme-miR-982-5p, dme-miR-982-3p, and miR-984-5p did not show a significant difference between Wolbachia-infected and uninfected fly testes (Fig. 5a, c and d). Again, we did not detect the expression level of dme-miR-983-3p in testes (data not shown).

qRT-PCR confirmation of differentially expressed miRNAs and their targets in adult gonads of Drosophila melanogaster. Dmel T: D. melanogaster treated with tetracycline (without Wolbachia); Dmel wMel: D. melanogaster infected with wMel Wolbachia. Red color indicates upregulation; Blue color indicates downregulation; Black color indicates not significantly different. A two-tailed Student’s t-test was performed to compare the significance between Wolbachia-infected and uninfected fly ovaries and testes of candidate miRNAs and their target genes. “/” represents the relative value; Bars indicate standard error, “*” and “**” indicate significant differences with p < 0.05 and p < 0.01, respectively. df = 4, n = 3
Target gene prediction of differentially expressed miRNAs
To further determine the biological functions of the differentially expressed miRNAs, miRanda was used to predict the target genes of these seven miRNAs. When target genes of differentially expressed miRNAs were further subjected to KEGG pathway analysis, the 20 most enriched pathways included phosphatidylinositol signaling, inositol phosphate metabolism, the Wnt signaling pathway, and the JAK-STAT signaling pathway (Fig. 6, Additional file 4). The GO annotation enrichment displayed that metabolic process, development process, and response to stimulus were included in the biological processes (Additional file 5).

The 20 most enriched KEGG pathways based on target genes of differentially expressed miRNAs in Wolbachia-infected and uninfected adult ovaries. The x-axis is the rich factor, and the y-axis is the pathway terms. The size of each point represents the number of genes enriched in a particular pathway. The larger the rich factor and the smaller the q-value, the more significant the degree of enrichment
Association analysis of Wolbachia infection-related miRNAs and mRNAs
In general, miRNAs have multiple target genes. The differentially expressed target genes that may be regulated by these seven Wolbachia-responsive miRNAs are presented in Table 4. qRT-PCR was performed to further examine how these target genes are associated with their Wolbachia-responsive miRNAs in ovaries and testes and thus with CI. The miRNAs dme-miR-982-5p and dme-miR-318-3p targeted the same gene, sosie, which is involved in ovarian follicle cell migration [27]. qRT-PCR results were consistent with RNA-seq analyses that sosie was significantly downregulated in fly ovaries due to Wolbachia infection, indicating that sosie is negatively regulated by these two miRNAs (Fig. 5a and e). The gene blanks, required for postmeiotic spermiogenesis [28], was positively regulated by dme-miR-983-5p in ovaries and negatively regulated in testes (Fig. 5b). The miRNA dme-miR-982-3p targeted three genes: nompC, Mal-A7, and tut. As shown in Fig. 2, nompC did not show a significant difference in expression between Wolbachia-infected and uninfected fly ovaries and testes. In contrast, Mal-A7 and tut were confirmed by qRT-PCR to be postively regulated by dme-miR-982-3p in fly ovaries (Fig. 5c). Mal-A7 was also significantly upregulated in Wolbachia-infected testes, though there was no difference in the expression of this miRNA in testes (Fig. 5c). The miRNA dme-miR-318-3p was significantly upregulated in ovaries but downregulated in testes due to Wolbachia infection. Its target Mlc2 was negatively regulated by dme-miR-318-3p in both ovaries and testes, exhibited downregulation in ovaries but upregulation in testes when compared to paired gonads without Wolbachia (Fig. 5e). In addition to Mlc2 and sosie, the third target of dme-miR-318-3p is Oamb, which showed no difference in expression in both ovaries and testes (data not shown). The expression of dme-miR-956-3p was not significantly changed by Wolbachia infection in ovaries, while its targets CG42324 and nolo were upregulated and downregulated, respectively, consistent with our RNA-seq data (Fig. 5f, Additional file 1). The third target gene of dme-miR-956-3p is tipE, which did not exhibit significant differences in both ovaries and testes in the presence and absence of Wolbachia (data not shown).
Discussion
For years, the molecular mechanisms of CI induced by Wolbachia remained unclear. Previous studies focusing on Wolbachia-infected males revealed a series of data on host gene expression changes, which may help to elucidate the possible molecular mechanisms of CI [11, 18,19,20,21]. Nevertheless, how Wolbachia-infected eggs may rescue the CI defect caused by Wolbachia-modified sperm and nullify embryonic death, and what makes the ovary a suitable place for Wolbachia to survive and transmit have not yet been elucidated. Therefore, we compared the transcriptional profiles including sRNAs between Wolbachia-infected and uninfected ovaries of D. melanogaster.
By using RNA-seq, we identified 149 differentially expressed genes in fly ovaries due to Wolbachia infection, of which 77.85% (116) showed significant upregulation. This generally altered gene expression pattern caused by Wolbachia infection is the reverse of that found in our previous work on the male aspect [20], where we identified 83 differentially expressed proteins by comparative proteomics in the spermatheca and seminal receptacle from uninfected females mated with Wolbachia-infected or uninfected males, and 71.08% (59) of them showed downregulation in the presence of Wolbachia [20]. Our findings broadly support the titration-restitution model proposed by Poinsot et al. to explain “modify/rescue” mechanism of CI. According to this model, Wolbachia may titrate-out some essential molecules from the sperm, and the same strain of Wolbachia in the egg can restore these critical factors, allowing embryos to develop normally [8].
Some studies have demonstrated that some genes potentially related to CI showed the opposite expression pattern in Wolbachia-infected male and female hosts. For instance, Ance and Hira were both upregulated in infected females and downregulated in infected males of Drosophila, which might be linked to CI expression [10, 11]. In the present study, we found a number of DEGs that were associated with cellular processes by influencing the expression of genes involved in transcription and translation. For example, RpL22-like (coding for ribosome protein L22e), Ptx1 (coding for paired-type homeobox protein), blanks (coding for siRNA binding protein and associated with regulation of chromatin silencing), and scro (coding for one of the homeobox proteins) were all upregulated in the presence of Wolbachia. This is consistent with the case of Encarsia suzannae infection with Cardinium strain cEper1, which also can induce CI in its insect hosts, where the authors found upregulation of several female-biased genes encoding ribosomal proteins [29], suggesting increased general translational activity in female hosts. Notably, RpL22-like and blanks were significantly downregulated in the testes of D. melanogaster due to Wolbachia infection (Fig. 2b and 5b). This altered expression pattern is consistent with the work with Ance [10] and Hira [11] and also matches the “titration-restitution” model for CI [8].
In this study, we also identified multiple differentially expressed genes involved in immunity in ovaries between Wolbachia-infected and uninfected D. melanogaster, most of which were downregulated including AttC (coding for Attacin-C) and Def (coding for Defensin). The altered expression of these two genes caused by Wolbachia infection was further confirmed by qRT-PCR in both ovaries and testes. The expression levels of both AttC and Def were significantly decreased in ovaries but increased in testes. The decreased expression of immune-related genes in ovaries may reflect a reduced pathogenicity caused by the symbiont and an increased tolerance of the host to Wolbachia, thus offering the benefit of maximum proliferation and transmission of the bacteria. On the other hand, the reduced immune response may also allow the infected eggs to fit the modified sperm once fertilization occurs, so as to ensure normal embryogenesis. This altered expression pattern is generally opposed to that in larval testes [18]. By microarray analyses, we previously revealed that most of the immune-associated genes were upregulated in the 3rd instar larval testes in the presence of Wolbachia [18]. Again, the opposite expression pattern between ovaries and testes of these immune genes is also in accord with the “titration-restitution” model [8].
Previous work has demonstrated that miRNAs could function in Wolbachia/host interactions [30,31,32,33,34,35]. For instance, in Aedes aegypti, the wMelpop-CLA strain of Wolbachia can alter aae-miR-2940 to increase metalloprotease gene expression for self-maintenance in mosquito hosts [30]. In insects, many lines of evidence revealed that miRNAs could operate in ovarian development [36,37,38,39,40]. Here, we reported for the first time that Wolbachia could modulate miRNAs of host ovaries. Upregulation of both dme-miR-318-3p and dme-miR-982-5p may ensure downregulation sosie in response to Wolbachia infection. The involvement of sosie in the coordination of migrations of ovarian border cells and outer follicle cells during mid-oogenesis has been demonstrated [27]. Interestingly, a recent work discovered a novel Wolbachia tropism (the polar cells) in Drosophila ovary, which may facilitate efficient vertical transmission of the endosymbiont [41]. During stage 9 of oogenesis, the anterior polar cells, along with a few surrounding border cells, invasively migrate through the nurse cells and towards the oocyte. This migration provides an adequate opportunity for Wolbachia to traverse from polar cells to the germline [41]. Downregulation of sosie may slow the migration of ovarian border cells and outer follicle cells thus aid Wolbachia in traversing into the germ line during oogenesis of Drosophila. Moreover, the opposite expression pattern of dme-miR-318-3p in ovaries (upregulated) and testes (downregulated) caused by Wolbachia infection is in accordance with its target Mlc2 expression pattern. Mlc2 codes for myosin light chain 2, which is normally involved in myofibril assembly. Recently, Zhang et al. found Mlc2 could also act as a transcription factor that can regulate the expression of nicotinamide adenine dinucleotide phosphate oxidase 2 to enhance oxidative stress in a phosphorylation-dependent manner [42]. The consistently altered expression patterns of dme-miR-318-3p and Mlc2 in response to Wolbachia suggest that miRNA pathways are involved in the induction and rescue of CI. And once again, the reverse altered expression patterns of dme-miR-318-3p and its target Mlc2 in ovaries and testes due to Wolbachia infection are consistent with the “titration-restitution” hypothesis [8].
Among those significantly upregulated genes, a large number are involved in metabolism processes and transport. These are consistent with findings in the previous experimental literature [18, 43, 44]. Caragata et al. found that numbers of genes encoding digestive enzymes, such as serine proteases or trypsin, showed increased expression in female Aedes fluviatilis in the presence of native wFlu Wolbachia [45]. In this research, we found that CG10659 (coding Acyl-CoA N-acyltransferase) displayed upregulation in the ovary. However, it did not show significant change in expression in the testis in response to Wolbachia infection (Fig. 2). This indicates that the increase in CG10659 expression resulting from Wolbachia infection is ovary-specific. CG32054, coding for a general substrate transporter, was also upregulated in Wolbachia-infected Drosophila ovaries but did not significantly alter due to Wolbachia infection in testes. These are consistent with our previous microarray analyses of the transcriptional profiles of D. melanogaster larval testes with and without wMel Wolbachia [18], suggesting that there might be some materials specially required in host ovaries for bacterial survival and propagation. This is not in accord with titration-restitution hypothesis. Instead, it may reflect a special requirement for Wolbachia propagation through the hosts’ eggs. The Drosophila ovary proteome and the female mosquito transcriptome also showed widespread upregulation of gene products involved in the transport and metabolism of amino acid, carbohydrate and lipid transporters [23, 45]. The genome sequence of the wMel strain showed that Wolbachia do not contain the complete set of metabolic pathways present in free-living bacteria [46]. Therefore, Wolbachia may not only upregulate the metabolism of their hosts for cannibalization of host resources, but also depend on a large assortment of transporters to obtain sufficient energy and critical components to promote their own proliferation and propagation [45,46,47].
Conclusions
In this study, mRNAs and miRNAs in fly ovaries with and without Wolbachia were compared. Our analyses revealed 149 mRNAs and seven miRNAs that exhibit significant changes in expression due to Wolbachia infection. These genes are involved in metabolism, transportation, oxidation-reduction, development, and other functions, suggesting a wide effect of Wolbachia on their female hosts. The differentially expressed genes are generally varied in opposite directions in male versus female flies in the presence of Wolbachia, which strongly supports the “titration-restitution” model for explaining CI mechanism. Furthermore, genes related to oxidation and reduction, as well as immunity, are also altered in fly ovaries by Wolbachia infection, reflecting a protection for host ovaries, thus ensuring the maximum proliferation and propagation of the bacteria. Our data provide insights into mechanisms of Wolbachia/host interactions, especially the Wolbachia-induced CI and Wolbachia dependence on host ovaries.
Methods
Fly stocks and rearing
Drosophila melanogaster flies were reared on a standard cornmeal- and molasses-based diet. Stocks were maintained at a constant temperature of 25 °C and under non-crowded conditions (approximate 200 eggs per 50-ml vial of media in 150-ml conical flasks) [48]. All flies were kept on a 12-h light/dark cycle. The D. melanogaster infected with wMel Wolbachia, referred to as Dmel wMel, was a gift from Professor Scott O’Neill at Monash University, Australia. Wolbachia-uninfected lines from Dmel wMel, referred to as Dmel T, were subsequently generated through tetracycline treatment as previously described [49] and verified to be Wolbachia-free by PCR using Wolbachia surface protein (wsp) primers (Additional file 6). The cured flies were reared on normal tetracycline-free medium for at least six generations to eliminate any influences of residual tetracycline before they were used in experiments [50].
RNA preparation
Eighty pairs of ovaries for each replicate were dissected from 4-day-old virgin females of Dmel T and Dmel wMel flies, respectively. RNA preparation and sequencing were performed by Novogene Bioinformatics Technology Co., Ltd. (Beijing, China). Briefly, total RNA was extracted using TRIzol reagent following the manufacturer’s instructions. Total RNA integrity was determined using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, USA), and concentration was measured using the Qubit RNA Assay Kit in the Qubit 2.0 Fluorometer (Life Technologies, CA, USA).
RNA sequencing
Sequencing libraries were prepared using NEBNext® Ultra™ RNA Library Prep Kit for Illumina (NEB, USA) according to the manufacturer’s recommendations. Briefly, mRNA was isolated from total RNA using Oligo-dT beads and then fragmentation was performed using NEBNext First Strand Synthesis Reaction Buffer (5X). First-strand cDNA was synthesized with random hexamer primers and M-MuLV Reverse Transcriptase. Second-strand cDNA was subsequently synthesized with DNA polymerase I and RNase H. Double-stranded cDNA fragments were purified with the AMPure XP system (Beckman Coulter, Beverly, USA) and adenylated at the 3′ ends. The NEBNext Adaptor with a hairpin loop structure was ligated to prepare for hybridization. The adaptor-ligated cDNA was used as a template to carry out PCR with Phusion High-Fidelity DNA polymerase and Universal PCR primers. Then the PCR products were purified with the AMPure XP system, and library quality was determined on the Agilent Bioanalyzer 2100 system. Finally, the cDNA library preparations were sequenced on an Illumina HiSeq 4000 platform and 150 bp paired-end reads were produced. Clean reads were obtained by removing reads containing adapter, ambiguous reads (the ratio of reads with poly N was greater than 10%, N indicated that base cannot be determined) and low-quality reads (The number of bases whose mass < =20 accounted for more than 50% of the total read length).
Differential gene expression calculation and pathway analysis
Differentially expressed genes were selected based on a fold change > = 2 and a p value < 0.05 with three biological replicates. KEGG pathway (Kyoto Encyclopedia of Genes and Genomes) enrichment analysis of DEGs was performed using KOBAS [51]. Gene Ontology (GO) enrichment analysis of differentially expressed genes was performed by using the GOseq R package.
Small RNA library construction for Illumina sequencing
Sequencing libraries were carried out using NEBNext Multiplex Small RNA Library Prep Set for Illumina (NEB, USA) following the manufacturer’s protocols. Briefly, the NEB 3′ adaptor was ligated to the 5′ and 3′ ends of miRNA using T4 ligase. The first-strand cDNA was synthesized using M-MLV Reverse Transcriptase. PCR amplification was conducted using LongAmp Taq 2X Master Mix. The PCR products were purified and then assessed on the Agilent Bioanalyzer 2100 system. At last, the cDNA library constructs were sequenced on an Illumina HiSeq 2500 platform and 50-bp single-end reads were generated. Clean reads were obtained by removing reads with 5′ primer contaminants, without 3′ primers or the insert tag, ambiguous reads (the ratio of reads with poly N was greater than 10%, N indicated that base cannot be determined), poly A/T/G/C reads and low-quality reads (The number of bases whose mass < =5 accounted for more than 50% of the total read length). Differentially expressed miRNAs were selected based on a fold change > = 2 and a p value < 0.05 with three biological replicates.
Target gene prediction and functional enrichment
Candidate targets of differentially expressed miRNAs were predicted by miRanda [52]. GO enrichment analysis was used to target gene candidates of differentially expressed miRNAs. KOBAS [51] was used to test the statistical enrichment of the target gene candidates in the KEGG pathways.
Quantitative reverse transcriptase PCR (qRT-PCR)
Forty pairs of ovaries for each biological replicate were dissected from 4-day-old virgin females of Dmel T and Dmel wMel flies, respectively. Sixty pairs of testes for each biological replicate were dissected from 1-day-old males of Dmel T and Dmel wMel flies, respectively. The qRT-PCR experiments were conducted using a CFX connect™ real-time system (BioRad). Total RNA was extracted using TRIzol (Invitrogen). DNA contamination was digested with RNase-free DNase I (Takara). The first-strand cDNA was synthesized from about 2 μg of total RNA using M-MLV reverse transcriptase (Invitrogen) and Oligo dT18 primer (Takara) at 37 °C for 50 min. Specific primers were designed based on sequences from the FlyBase database (Additional file 6). qPCR was performed with a Platinum SYBR Green qPCR superMix (Takara). Each reaction mixture contained 10 μl of 2× SYBR premix, 0.3 μl of forward and reverse primer (10 μM), respectively, 2 μl of cDNA template diluted 5-fold with deionized H2O, and deionized H2O to a final volume of 20 μl. The cycling program was 95 °C for 2 min, followed by 40 cycles of 95 °C for 10 s, 56–58 °C (based on various primers) for 20 s and 72 °C for 20 s, and then a melting curve was constructed from 55 °C to 98 °C. The relative expression of each gene was calibrated against the reference gene (rp49) using 2-ΔCT (ΔCT = CT, target gene - CT, rp49). Three biological replicates and two technical replicates for each biological replicate were performed.
miRNA expression patterns
The experiments were performed on a CFX connect™ real-time system (BioRad) with three biological replicates and two technical replicates. Forty pairs of 4-day-old virgin female ovaries and sixty pairs of 1-day-old male testes were dissected for each biological replicate. Total RNA was extracted with a miRNeasy extraction kit (TIANGEN catalog no. DP501) together with an RNase-Free DNase set. RNA quality was estimated with a Nanodrop spectrophotometer and by agarose gel electrophoresis. The expression of miRNA was checked by using the SYBR Prime-Script miRNA RT-PCR Kit (TIANGEN catalog no. FP401). Total RNA (2 μg) was reverse transcribed, and 30 ng cDNA products were added to a 20-μL quantification system. The primers used in this study are shown in Additional file 6. The reactions were incubated at 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s and 65 °C for 34 s. A dissociation curve was obtained to ensure that only one product was amplified after the amplification phase. The relative expression of each miRNA was normalized against the reference gene (U6 snRNA) using 2-ΔCT (ΔCT = CT, target miRNA - CT, U6 snRNA).
Statistical analyses
For all experiments, a two-tailed Student’s t-test was performed. All data were represented as the means ± standard error (SE, n = 3) and were graphed by GraphPad Prism 6.
Availability of data and materials
The datasets generated and/or analyzed during the current study are publicly available at NCBI-SRA (www.ncbi.nlm.nih.gov/sra) accession: SRP136211.
Abbreviations
- CYP:
-
Cytochrome P450
- DEGs:
-
Differentially expressed genes
- Dmel T:
-
Drosophila melanogaster treated with tetracycline (without Wolbachia)
- Dmel wMel:
-
Drosophila melanogaster infected with wMel Wolbachia
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PIWI Interacting RNA:
-
piRNA
- RNA-seq:
-
RNA sequencing
- sRNA-seq:
-
Small RNA sequencing
References
Werren JH, Baldo L, Clark ME. Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol. 2008;6:741–51.
Zug R, Hammerstein P. Still a host of hosts for Wolbachia: analysis of recent data suggests that 40% of terrestrial arthropod species are infected. PLoS One. 2012;7(6):e38544.
Serbus LR, Casperlindley C, Landmann F, Sullivan W. The genetics and cell biology of Wolbachia-host interactions. Annu Rev Genet. 2008;42:683–707.
Yen JH, Barr AR. New hypothesis of the cause of cytoplasmic incompatibility in Culex pipiens L. Nature. 1971;232(5313):657–8.
Tram U, Fredrick K, Werren JH, Sullivan W. Paternal chromosome segregation during the first mitotic division determines Wolbachia-induced cytoplasmic incompatibility phenotype. J Cell Sci. 2006;119(17):3655–63.
Bonneau M, Landmann F, Labbé P, Justy F, Weill M, Sicard M. The cellular phenotype of cytoplasmic incompatibility in Culex pipiens in the light of cidB diversity. PLoS Pathog. 2018;14(10):e1007364.
Bourtzis K, Dobson SL, Braig HR, O'neill SL. Rescuing Wolbachia have been overlooked. Nature. 1988;391(6670):852–3.
Poinsot D, Charlat S, Mercot H. On the mechanism of Wolbachia-induced cytoplasmic incompatibility: confronting the models with the facts. Bioessays. 2003;25(3):259–65.
Bossan B, Koehncke A, Hammerstein P. A new model and method for understanding Wolbachia-induced cytoplasmic incompatibility. PLoS One. 2011;6(5):e19757.
Xi Z, Gavotte L, Xie Y, Dobson SL. Genome-wide analysis of the interaction between the endosymbiotic bacterium Wolbachia and its Drosophila host. BMC Genomics. 2008;9(1):1.
Zheng Y, Ren PP, Wang JL, Wang YF. Wolbachia-induced cytoplasmic incompatibility is associated with decreased Hira expression in male Drosophila. PLoS One. 2011;6(4):e19512.
Beckmann JF, Markowski TW, Witthuhn BA, Fallon AM. Detection of the Wolbachia-encoded DNA binding protein, HU beta, in mosquito gonads. Insect Biochem Mol Biol. 2013;43(3):272–9.
Beckmann JF, Fallon AM. Detection of the Wolbachia protein WPIP0282 in mosquito spermathecae: implications for cytoplasmic incompatibility. Insect Biochem Mol Biol. 2013;43(9):867–78.
Beckmann JF, Ronau JA, Hochstrasser M. A Wolbachia deubiquitylating enzyme induces cytoplasmic incompatibility. Nat Microbiol. 2017;2:17007.
Ote M, Ueyama M, Yamamoto D. Wolbachia protein TomO targets nanos mRNA and restores germ stem cells in Drosophila sex-lethal mutants. Curr Biol. 2016;26(17):2223–32.
LePage DP, Metcalf JA, Bordenstein SR, On J, Perlmutter JI, Shropshire JD, et al. Prophage WO genes recapitulate and enhance Wolbachia-induced cytoplasmic incompatibility. Nature. 2017;543(7644):243–7.
Shropshire JD, On J, Layton EM, Zhou H, Bordenstein SR. One prophage WO gene rescues cytoplasmic incompatibility in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2018;115(19):4987–91.
Zheng Y, Wang JL, Liu C, Wang CP, Walker T, Wang YF. Differentially expressed profiles in the larval testes of Wolbachia infected and uninfected Drosophila. BMC Genomics. 2011;12(1):595.
Liu C, Wang JL, Zheng Y, Xiong EJ, Li JJ, Yuan LL, et al. Wolbachia-induced paternal defect in Drosophila is likely by interaction with the juvenile hormone pathway. Insect Biochem Mol Biol. 2014;49:49–58.
Yuan LL, Chen XL, Zong Q, Zhao T, Wang JL, Zheng Y, et al. Quantitative proteomic analyses of molecular mechanisms associated with cytoplasmic incompatibility in Drosophila melanogaster induced by Wolbachia. J Proteome Res. 2015;14:3835–47.
Ju JF, Hoffmann AA, Zhang YK, Duan XZ, Guo Y, Gong JT, et al. Wolbachia-induced loss of male fertility is likely related to branch chain amino acid biosynthesis and iLvE in Laodelphax striatellus. Insect Biochem Mol Biol. 2017;85:11–20.
Landmann F, Orsi GA, Loppin B, Sullivan W. Wolbachia-mediated cytoplasmic incompatibility is associated with impaired histone deposition in the male pronucleus. PLoS Pathog. 2009;5(3):e1000343.
Christensen S, Dulzaides RP, Hedrick VE, Momtaz AJMZ, Nakayasu ES, Paul LN, et al. Wolbachia endosymbionts modify Drosophila ovary protein levels in a context-dependent manner. Appl Environ Microbiol. 2016;82(17):5354–63.
Reynolds KT, Hoffmann AA. Male age, host effects and the weak expression or non-expression of cytoplasmic incompatibility in Drosophila strains infected by maternally transmitted Wolbachia. Genet Res. 2002;80(2):79–87.
Huang R, Jaritz M, Guenzl P, Vlatkovic I, Sommer A, Tamir IM, et al. An RNA-seq strategy to detect the complete coding and non-coding transcriptome including full-length imprinted macro ncRNAs. PLoS One. 2011;6(11):e27288.
Ylla G, Piulachs MD, Belles X. Comparative analysis of miRNA expression during the development of insects of different metamorphosis modes and germ-band types. BMC Genomics. 2017;18(1):774.
Urwyler O, Cortinas-Elizondo F, Suter B. Drosophila sosie functions with βH-Spectrin and actin organizers in cell migration, epithelial morphogenesis and cortical stability. Biol Open. 2012;1(10):994–1005.
Gerbasi VR, Preall JB, Golden DE, Powell DW, Cummins TD, Sontheimer EJ. Blanks, a nuclear siRNA/dsRNA-binding complex component, is required for Drosophila spermiogenesis. Proc Natl Acad Sci U S A. 2011;108(8):3204–9.
Mann E, Stouthamer CM, Kelly SE, Dzieciol M, Hunter MS, Schmitz-Esser S. Transcriptome sequencing reveals novel candidate genes for Cardinium hertigii-caused cytoplasmic incompatibility and host-cell interaction. mSystems. 2017;2:e00141–17.
Hussain M, Frentiu FD, Moreira LA, O'Neill SL, Asgari S. Wolbachia uses host microRNAs to manipulate host gene expression and facilitate colonization of the dengue vector Aedes aegypti. Proc Natl Acad Sci U S A. 2011;108(22):9250–5.
Hussain M, O’Neill SL, Asgari S. Wolbachia interferes with the intracellular distribution of Argonaute 1 in the dengue vector Aedes aegypti by manipulating the host microRNAs. RNA Biol. 2013;10(12):1868–75.
Osei-Amo S, Hussain M, O’Neill SL, Asgari S. Wolbachia-induced aae-miR-12 miRNA negatively regulates the expression of MCT1 and MCM6 genes in Wolbachia-infected mosquito cell line. PLoS One. 2012;7(11):e50049.
Zhang G, Hussain M, O’Neill SL, Asgari S. Wolbachia uses a host microRNA to regulate transcripts of a methyltransferase, contributing to dengue virus inhibition in Aedes aegypti. Proc Natl Acad Sci U S A. 2013;110(25):10276–81.
Mayoral JG, Etebari K, Hussain M, Khromykh AA, Asgari S. Wolbachia infection modifies the profile, shuttling and structure of microRNAs in a mosquito cell line. PLoS One. 2014;9(4):e96107.
Rong X, Zhang YK, Zhang KJ, Hong XY. Identification of Wolbachia-responsive microRNAs in the two-spotted spider mite, Tetranychus urticae. BMC Genomics. 2014;15(1):1122.
Fu X, Li T, Chen J, Dong Y, Qiu J, Kang K, et al. Functional screen for microRNAs of Nilaparvata lugens reveals that targeting of glutamine synthase by miR-4868b regulates fecundity. J Insect Physiol. 2015;83:22–9.
Kugler JM, Chen YW, Weng R, Cohen SM. miR-989 is required for border cell migration in the Drosophila ovary. PLoS One. 2013;8(7):e67075.
Hao J, Luo J, Chen Z, Ren Q, Guo J, Liu X, et al. MicroRNA-275 and its target Vitellogenin-2 are crucial in ovary development and blood digestion of Haemaphysalis longicornis. Parasites Vectors. 2017;10(1):253.
Zhang Y, Zhao B, Roy S, Saha TT, Kokoza VA, Li M, et al. microRNA-309 targets the Homeobox gene SIX4 and controls ovarian development in the mosquito Aedes aegypti. Proc Natl Acad Sci U S A. 2016;113(33):E4828–36.
Lucas KJ, Zhao B, Liu S, Raikhel AS. Regulation of physiological processes by microRNAs in insects. Curr Opin Insect Sci. 2015;11:1–7.
Kamath AD, Deehan MA, Frydman HM. Polar cell fate stimulates Wolbachia intracellular growth. Development. 2018; dev.158097.
Zhang YS, Liu B, Luo XJ, Li TB, Zhang JJ, Peng JJ, et al. Nuclear cardiac myosin light chain 2 modulates NAPHG oxidase 2 expression in myocardium: a novel function beyond muscle contraction. Basic Res Cardiol. 2015;110(4):1–17.
Molloy JC, Sommer U, Viant MR, Sinkins SP. Wolbachia modulates lipid metabolism in aedes albopictus mosquito cells. Appl Environ Microbiol. 2016;82(10):3109–20.
Geoghegan V, Stainton K, Rainey SM, Ant TH, Dowle AA, Larson T, et al. Perturbed cholesterol and vesicular trafficking associated with dengue blocking in Wolbachia-infected Aedes aegypti cells. Nat Commun. 2017;8(1):526.
Caragata EP, Pais FS, Baton LA, Silva JBL, Sorgine MHF, Moreira LA. The transcriptome of the mosquito Aedes fluviatilis (Diptera: Culicidae), and transcriptional changes associated with its native Wolbachia infection. BMC Genomics. 2017;18(1):6.
Wu M, Sun LV, Vamathevan J, Riegler M, Deboy R, Brownlie JC, et al. Phylogenomics of the reproductive parasite Wolbachia pipientis wMel: a streamlined genome overrun by mobile genetic elements. PLoS Biol. 2004;2(3):E69.
Cho KO, Kim GW, Lee OK. Wolbachia bacteria reside in host Golgi-related vesicles whose position is regulated by polarity proteins. PLoS One. 2011;6(7):e22703.
Yamada R, Floate KD, Riegler M, O'Neill SL. Male development time influences the strength of Wolbachia-induced cytoplasmic incompatibility expression in Drosophila melanogaster. Genetics. 2007;177(2):801–8.
Hoffmann AA, Turelli M, Simmons GM, Simmons GM. Unidirectional incompatibility between populations of Drosophila simulans. Evolution. 1986;40(4):692–701.
Chrostek E, Marialva MSP, Esteves SS, Weinert LA, Martinez J, Jiggins FM, et al. Wolbachia variants induce differential protection to viruses in Drosophila melanogaster: a phenotypic and phylogenomic analysis. PLoS Genet. 2013;9:e1003896.
Mao X, Cai T, Olyarchuk JG, Wei L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21(19):3787–93.
Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003;5:R1.
Acknowledgments
Authors thank Professor Scott O’Neill (Monash University, Australia) for providing Dmel wMel flies.
Funding
This work was funded by the grant from the National Natural Science Foundation of China (No. 31672352). The funding body had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Experimental concept and design: YZ and YFW. Research experimentation: ZH, WJY and YF. Analysis of all results: ZH, BM and YFW. Manuscript writing and figures: ZH, BM and YFW. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Differentially expressed genes in ovaries of D. melanogaster induced by Wolbachia infection. (XLS 57 kb)
Additional file 2:
The 20 most enriched KEGG pathways from enrichment analysis of differentially expressed genes in ovaries of D. melanogaster induced by Wolbachia infection. (XLS 30 kb)
Additional file 3:
GO analysis of differentially expressed genes in ovaries of D. melanogaster induced by Wolbachia infection. (XLS 53 kb)
Additional file 4:
The 20 most enriched KEGG pathways based on target genes of differentially expressed miRNAs in ovaries of D. melanogaster induced by Wolbachia infection. (XLS 33 kb)
Additional file 5:
GO analysis of target genes of differentially expressed miRNAs in ovaries of D. melanogaster induced by Wolbachia infection. (XLS 305 kb)
Additional file 6:
List of primers used for qRT-PCR or PCR analysis in this work. (XLS 36 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
He, Z., Zheng, Y., Yu, WJ. et al. How do Wolbachia modify the Drosophila ovary? New evidences support the “titration-restitution” model for the mechanisms of Wolbachia-induced CI. BMC Genomics 20, 608 (2019). https://doi.org/10.1186/s12864-019-5977-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-5977-6