
Abstract
Background
The behavioural transition from nurses to foragers in honey bees is known to be affected by intrinsic and extrinsic factors, including colony demography, hormone levels, brain chemistry and structure, and gene expression in the brain. However, the molecular mechanism underlying this behavioural transition of honey bees is still obscure.
Results
Through RNA sequencing, we performed a comprehensive analysis of lncRNAs and mRNAs in honey bee nurses and foragers. Nurses and foragers from both typical colonies and single-cohort colonies were used to prepare six libraries to generate 49 to 100 million clear reads per sample. We obtained 6863 novel lncRNAs, 1480 differentially expressed lncRNAs between nurses and foragers, and 9308 mRNAs. Consistent with previous studies, lncRNAs showed features distinct from mRNAs, such as shorter lengths, lower exon numbers, and lower expression levels compared to mRNAs. Bioinformatic analysis showed that differentially expressed genes were mostly involved in the regulation of sensory-related events, such as olfactory receptor activity and odorant binding, and enriched Wnt and FoxO signaling pathways. Moreover, we found that lncRNAs TCONS_00356023, TCONS_00357367, TCONS_00159909 and mRNAs dop1, Kr-h1 and HR38 may play important roles in behavioural transition in honey bees.
Conclusion
This study characterized the expression profile of lncRNAs in nurses and foragers and provided a framework for further study of the role of lncRNAs in honey bee behavioural transition.
Similar content being viewed by others


Background
The Western honey bee, Apis mellifera L., a social insect and a good agricultural pollinator, possesses a remarkable trait: the behavioural transition from inhive tasks to outside tasks. In a typical colony, young worker honey bees (nurses) take care of the brood; approximately one week later, worker bees change to new roles, such as storing and processing food in colonies; when they are three weeks of age, worker bees begin to forage for honey, pollen, propolis or water outside of the hive [1]. Foragers can change back to be nurses under certain conditions [2]. Such behavioural plasticity has attracted much research attention, and a number of factors have been reported to be associated with it: colony demography [3], hormone levels [4], exocrine gland activity [5], brain chemistry and structure [6], circadian rhythms [7], and gene expression in the brain [8]. Non-coding RNAs (ncRNA) are those RNAs not involved in coding; they include small RNAs (18–35 nucleotides, nt), and long non-coding RNAs (lncRNA), with lengths > 200 nt. lncRNAs can be classified as natural antisense transcripts, long intronic non-coding RNAs, or long intergenic non-coding RNAs (lincRNAs) according to their genomic position [9]. In recent years, lncRNAs have been confirmed to play roles in many biological processes, such as cell differentiation and development, immune responses and tumourigenesis [10,11,12]. A most recent study showed that most lncRNAs were dysregulated in a tumour-specific manner and synergistically dysregulated cancer pathways in multiple tumour contexts [13]. Many Drosophila melanogaster lncRNAs have been found to be associated with X inactivation [14, 15], behaviour [16, 17], and neuronal disease [18].
lncRNAs in honey bees have also been observed to function in developmental processes. Both AncR-1 and Ks-1 have abundant expression in the brain and accumulate in the nucleus, showing their potential role in the regulation of neural function in honey bees [19, 20]. Kakusei in the nucleus is activated in a subset of neurons in the brains of dancing foraging bees, and Nb-1 is involved in regulating octopamine and juvenile hormone release during the behavioural transition from nursing to foraging [21, 22], suggesting that they are involved in the regulation of behaviours in honey bees. Jayakodi et al. (2015) identified lincRNAs specifically associated with viral diseases in honey bees, and they were preferentially expressed in ovary tissue [23]. Chen et al. (2017) further observed dramatic expression changes of coding and noncoding RNAs, suggesting that they play a critical role in oviposition in honey bee queens [24]. A context-dependent transcription of one lncRNA encoding an anti-sense transcript of lysosomal alpha-mannosidase in the honey bee has been shown to be linked to DNA methylation [25]. However, there are no reports about the role of lncRNA in the division of labour in honey bees.
In this study, we used RNA-seq to detect the profile of lncRNA in the heads of nurses and foragers from typical colonies and obtained many differentially expressed transcripts. In addition, to separate whether the nurse-forager difference was due to age or task performance, we compared lncRNA profiles of bees in typical colonies with those of bees from single-cohort colonies.
Results
lncRNAs identified from RNA-seq
Total RNA from the heads of six honey bee groups (TC_N, TC_F, SCC_YN, SCC_YF, SCC_ON, SCC_OF, see methods for explanation) was isolated and sequenced. Approximately 50 to 102 million raw reads and 49 to 100 million clean reads per sample were obtained by RNA-Seq (Additional file 1). The sequence reads were mapped with a reference annotation. Approximately 82.51 to 86.79% of the reads were mapped to mRNAs, depending on the group (Additional file 2). For ncRNAs, 1.17 to 2.08% of the sample reads were mapped (Additional file 2). We identified 7470 novel lncRNAs after using Cufflinks (v2.1.1) [26] and Scripture (beta2) [27]. Then, 6863 putative non-coding transcripts were obtained using Coding Potential Calculator (CPC) and Pfam-scan (PFAM) (Fig. 1). We also obtained 22,203 transcripts of mRNAs, with 21,596 transcripts mapped to the reference genome, with the remaining coding transcripts (607) not annotated.

Identification of non-coding lncRNAs by using PFAM and CPC. A total of 6863 non-coding transcripts were selected using two software programs evaluating protein-coding transcripts to remove putative protein-coding transcripts
Comparative features of mRNAs and lncRNAs
A total of 21,596 mRNAs and 6863 lncRNAs were obtained from the honey bee heads (all samples combined). Most lncRNAs contained one to two exons, which was significantly different from that of mRNAs (Chi-square test, df = 29, χ2 = 21,019.93, P < 0.0001, Fig. 2a), and there was a great divergence in the distribution of transcript length between mRNAs and lncRNAs (Chi-square test, df = 19, χ2 = 3701.49, P < 0.0001, Fig. 2b). Moreover, we found that most lncRNAs had significantly shorter ORFs ranging from 30 bp to 240 bp (Chi-square test, df = 30, χ2 = 22,413.34, P < 0.0001) compared to mRNAs (30 to > 900 bp) (Fig. 2c), and lncRNAs showed a significantly lower (T-test, P < 0.0001) expression level compared to mRNAs (Fig. 2d).

Exon number distribution of coding transcripts (mRNAs) and lncRNAs (a). Length distribution of 6863 new predicted lncRNAs (red) and 21,597 coding transcripts (blue) (b). ORF length distribution of mRNAs and lncRNAs (c). Expression level indicated by log10 (FPKM + 1) in the mRNAs and lncRNAs (d)
Differentially expressed genes between nurses and foragers
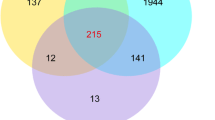
A total of 1480 differentially expressed lncRNAs and 9308 mRNAs were detected from pairwise nurse-forager comparisons in samples from typical colonies and single-cohort colonies using Cufflinks (v2.1.1) [26] (Table 1). The heatmaps of these genes are displayed in Additional file 3. A total of 449 upregulated and 37 downregulated lncRNAs were detected in the TC_F vs. TC_N comparison, 846 and 37 in the SCC_OF vs. SCC_ON comparison, and 368 and 83 in the SCC_YF vs. SCC_YN comparison, respectively. Additionally, 2925 upregulated and 581 downregulated mRNAs were detected in the TC_F vs. TC_N comparison, 6116 and 395 in the SCC_OF vs. SCC_ON comparison, and 2031 and 812 in the SCC_YF vs. SCC_YN comparison, respectively. As shown in Figs. 3, 52 differentially expressed lncRNAs and 645 differentially expressed mRNAs were common among the three contrasts (Additional file 4). We also found that 123 differentially expressed lncRNAs and 1293 differentially expressed mRNAs were common between the SCC_OF vs. SCC_ON and SCC_YF vs. SCC_YN comparisons (Additional file 5).

Gene expression profiles and number of differentially expressed genes for honey bees. Venn diagram of common differentially expressed genes (lncRNA and mRNA) among the three comparison groups
GO and KEGG analysis of target genes
We predicted the potential targets of lncRNAs by cis and trans regulation. A total of 3992 target genes (10 kb) and 9452 target genes (100 kb) were predicted in the cis role (data not shown). GO analysis showed that 20 significantly enriched GO terms (corrected p-value < 0.05) were detected in the TC_F vs. TC_N comparison and 15 terms were predicted in the SCC_YF vs. SCC_YN comparison, and most of the GO terms were related to regulation of sensory behaviour, such as sensory perception of smell, olfactory receptor activity and odorant binding (Fig. 4). However, there was no significant enrichment of GO terms for the SCC_OF vs. SCC_ON comparison. The most enriched pathways in the three comparisons of honey bees included “carbon metabolism”, “glycine, serine and threonine metabolism”, and the “phosphatidylinositol signalings system”. Among the top 20 enriched pathways, “Hippo signaling pathway - fly” and “Wnt signaling pathway” were the common pathways in the three comparison groups (Additional file 6).

The significant enrichment GO terms (corrected p value < 0.05) detected in the TC_F vs. TC_N (a) and SCC_YF vs. SCC_YN (b) comparisons (cis). Green bars represent molecular function terms; black bars represent cellular component terms; orange bars represent biological process terms
For the trans action of lncRNAs, 10,196 target genes were predicted. There were 118 significantly enriched GO terms (corrected p-value < 0.05) between the TC_F vs. TC_N comparison, 110 terms in the SCC_YF vs. SCC_YN and 46 terms in the SCC_OF vs. SCC_ON comparison (Additional file 7). These GO terms included a variety of molecular functions. We also found that “binding” and “protein binding” were the most common significantly enriched terms in each of the honey bee comparisons. The most enriched pathways in the three comparisons (TC_F vs. TC_N, SCC_YF vs. SCC_YN, SCC_OF vs. SCC_ON) of honey bees were the “endocytosis”, “ubiquitin mediated proteolysis”, and “FoxO signaling pathway”, respectively. Among the top 20 enriched pathways, the “Wnt signaling pathway” was the most common pathway in all three comparisons (Additional file 8).
Functional analysis of mRNA in honey bee samples
GO and KEGG enrichment analyses were also performed with differentially expressed mRNAs. In the TC_F vs. TC_N comparison, a total of 60, 24, and 62 GO terms were significantly enriched in the biological process, cell component, and molecular function, respectively. In the SCC_OF vs. SCC_ON comparison, 44, 11 and 45 GO terms were significantly enriched in the biological process, cell component, and molecular function, respectively. In the SCC_YF vs. SCC_YN comparison, 17, 14 and 37 were significantly enriched in the biological process, cell component, and molecular function, respectively (Additional file 9). The most enriched terms were associated with the gene expression and behaviour of honey bees. “Protein binding”, “binding”, “signaling”, “signal transduction”, “response to stimulus”, “Wnt signaling pathway”, “neuroactive ligand-receptor interaction”, “FoxO signaling pathway”, “notch signaling pathway”, and the “Hippo signaling pathway - fly” were common pathways among the top 20 enriched pathways in the three comparisons (Additional file 10). Interestingly, we observed that the “Wnt signaling pathway” was the most enriched pathway in all three comparisons.
Validation of selected lncRNAs and mRNAs
To validate the RNA-seq results, we chose three lncRNAs (TCONS_00357367, TCONS_00356023, TCONS_00159909) and three mRNAs (dop1, Kr-h1, HR38) for quantitative PCR (qPCR) (Table 2). The validated lncRNAs were significantly differentially expressed among the comparison groups, and the predicted target genes of these lncRNAs were previously shown to be associated with the division of labour in honey bees. The mRNAs selected were significantly differentially expressed among the three honey bee comparison groups. As shown in Fig. 5, TCONS_00357367, TCONS_00356023, dop1, Kr-h and HR38 had significantly higher expression in foragers than in nurses, which is consistent with our RNA-seq data. TCONS_00159909 had significantly higher expression in honey bee nurses than in foragers in the TC_F vs. TC_N and SCC_YF vs. SCC_YN comparisons but did not show significantly different expression between the SCC_ON and SCC_OF comparison (Fig. 5).

Validation of selected lncRNAs and mRNAs by using quantitative PCR. The relative expression levels of TCONS_00356023, TCONS_00357367, HR38, dop1, Kr-h1 in the heads of nurses and foragers from typical colonies (a); the relative expression levels of TCONS_00356023, TCONS_00357367, HR38, dop1, Kr-h1 in the heads of normal nurses and precocious foragers from single-cohort colonies (b); the relative expression levels of TCONS_00356023, TCONS_00357367, HR38, dop1, Kr-h1 in the heads of overaged nurses and normal-aged foragers from single-cohort colonies (c); the relative expression level of TCONS_00159909 in the heads of nurses and foragers from both typical colonies and single-cohort colonies. TCONS_00159909 expression was presented in a single figure, as its expression level was too low to be shown in the same plot as the other two lncRNAs (d). Blue represents nurses, and white represents foragers. Data were normalized to the housekeeping gene β-actin and are shown as the mean ± standard error, *P < 0.05, **P < 0.01
Discussion
In this study, RNA-seq was performed to build the lncRNA and mRNA profiles of honey bees. We obtained 6863 putative non-coding transcripts, 1480 differentially expressed lncRNAs, and 9308 differentially expressed mRNAs in the nurse vs. forager comparisons in bees from both typical colonies and single-cohort colonies. A total of 52 significantly differentially expressed lncRNAs and 645 such mRNAs were shared among the three comparisons (TC_F vs. TC_N, SCC_OF vs. SCC_ON and SCC_YF vs. SCC_YN, Fig. 3). Bioinformatic analysis showed that the “Wnt signaling pathway” may play an important role in the behavioural changes of honey bees.
lncRNA have been involved in essential biological processes, such as imprinting, gene regulation and dosage compensation, especially in mammals [28]. It has been reported that lncRNA regulates sleep behaviour, locomotor behaviour and immunity in Drosophila [29,30,31]. In this study, we found that several lncRNAs were actively involved in honey bee behavioural changes. First, lncRNAs TCONS_00207749 and TCONS_00207751 had lower expression in foragers than in nurses, and they both target the foraging gene (a cGMP-dependent protein kinase) in cis. The gene foraging was shown to have significantly higher expression in foragers than in nurses and plays an important role in regulating the behavioural transition from nursing to foraging [32]. Because of their inhibitory role towards foraging, their lower expression levels in foragers are consistent with the higher expression of foraging in foragers. Therefore, these two lncRNAs may be involved in modulating honey bee behaviour via modulating foraging. Furthermore, we found that lncRNA TCONS_00357367 was upregulated in foragers from both typical and single-cohort colonies (Additional file 4), while its target gene ACSF2 (acyl-CoA synthetase family member 2) in cis was downregulated in foragers in these colonies. Acyl-CoA synthetases (ACS) carry out a fundamental reaction in fatty acid metabolism [33], and dysregulation of fatty acid metabolism by disruption of ACS function in vivo can lead to neurodegenerative pathologies, which is evident in neuronal cells of Drosophila [34]. Foragers (typically older bees) had lower lipid amounts than did nurses (typically young bees) [35]. Here, we found that ACSF2 was significantly involved in the molecular function, catalytic activity, and metabolism processes. TCONS_00356023 was also upregulated in foragers in both typical and single-cohort colonies, and it targets Vg (vitellogenin) in cis. This is consistent with previous findings of lower Vg expression in foragers than in nurses [36] (Additional file 4). Early foraging onset can be induced by inhibition of vitellogenin mRNA (vg) production through RNAi [37]. Moreover, TCONS_00159909 was upregulated in nurses, and its predicted targets in cis and trans included major royal jelly proteins (mrjp1-mrjp9), yellow-e, yellow-e3, yellow-g, yellow-g2 and yellow-h. The MRJP protein family is evolved from the ancient Yellow protein family [38]. Mrjps were found to be expressed in the mushroom bodies of the honey bee brain. Mrjp1, coding for the most abundant protein of larval food, may regulate the learning ability of the honey bee [39, 40]. The expression of TCONS_00357367, TCONS_00356023 and TCONS_00159909 were further confirmed by qPCR (Fig. 5), the results of which were consistent with the RNA-seq data.
Over 1500 genes are differentially expressed in the brains of nurses and foragers [41]. Foraging and malvolio are among the presumably many genes that play a causal role in the division of labour of honey bees [32, 42]. In the present study, we found that several mRNAs were actively involved in honey bee behavioural changes. AmDOP1 was shown to be highly abundant in the soma of mushroom body intrinsic neurons of honey bees and was involved in signal processing of visual and olfactory information [43]. It has similar function to the Drosophila DAMB (dopamine receptor in mushroom bodies) gene, which plays key roles in arousal and memory [44]. In this study, we found that dop1 had a high expression in foragers but no expression in nurses in both typical and single-cohort colonies. We also found that dop1 was significantly enriched in the “signal transduction”, “signaling”, and “response to stimulus” terms and was related to the “neuroactive ligand-receptor interaction” pathway, suggesting its importance in the function of honey bee behaviour. Expression of the transcription factor Krüppel-homolog 1 (Kr-h1) was significantly higher in foragers than in nurses and is associated with cGMP-mediated changes in the brain that occur early in the transition to foraging behaviour [41]. Here, we found that Kr-h1 was upregulated in foragers in both typical and single-cohort colonies and involved in “signal transduction”, “signaling”, “response to stimulus”, and “binding” terms. A recent study showed that HR38 mediated 20-hydroxyecdysone regulating carbohydrate metabolism during mosquito reproduction [45]. In this study, we found that HR38 was upregulated in foragers in both typical and single-cohort colonies, which is similar to the results of the Khamis et al. (2015) study, which showed that HR38 had a higher expression in the brain of foragers compared to nurses and was concentrated in a subset of the mushroom body neurons of foragers [46]. The HR38-mediated pathway (ecdysteroid signaling) in the mushroom bodies was suspected to be involved in the division of labour by the workers [47]. Furthermore, the expression levels of dop1, Kr-h1, and HR38 were confirmed by qPCR in this study (Fig. 5), and were consistent with the RNA-seq data. These results suggest that these genes may play important roles in the honey bee behavioural transition.
Through bioinformatic analysis, we found that the “Wnt signaling pathway” was the most enriched pathway both in mRNAs and the target genes of lncRNAs. This suggests that Wnt may play a critical role in honey bee behavioural maturation. The honey bee genome has the same number of Wnt genes as that of Drosophila, and many features of Wnt signaling are conserved between the two species [48]. Wnt signaling is involved in embryogenesis and imaginal disc development in Drosophila [49]. Additionally, it can cross-talk with JH (juvenile hormone)-signaling by suppressing transcription of genes encoding for putative JH receptors to induce downregulation of Kr-h1expression in the early larval stages of Drosophila [50]. JH plays a critical role in honey bee development, including the regulation of division of labour [51]. Honey bees lacking the hormone would perform foraging later than normal bees [52]. Wnt signaling is one of the most crucial morphogens for development; during the maturation of the central nervous system its action is associated with the establishment and maintenance of synaptic structure and neuronal function [53]. Dysregulated Wnt signaling can lead to disorders of behaviour [54]. In this study, we found that TCONS_00357367 also targets the gene Pkc (protein kinase C), which was enriched in the Wnt signaling pathway, and it had significantly higher expression in foragers than in nurses. PKC had high expression in mushroom bodies and the antennal lobes, neuropils involved in the processing of sensory information and in learning [55]. In Drosophila inhibited PKC leads to a dissociation of their acquisition of learning and memory from their performance of a task [56]. Taken together, we deduce that the Wnt signaling pathway may be involved in the modulation of honey bee behaviour by regulating the neuronal function of the honey bee brain or that it may interact with the JH pathway to affect honey bee behaviours.
Conclusions
We first generated the expression profile of lncRNA in nurses and foragers by deep RNA-seq. Bioinformatic analysis showed that some lncRNAs and mRNAs were involved in important biological processes associated with honey bee behaviours, such as sensory perception of smell, olfactory receptor activity and odorant binding, and these lncRNAs may be involved in regulating the division of labour in honey bees by targeting mRNAs. Additionally, we found that “Wnt signaling pathway” may be involved in honey bee behavioural transition. As the research of lncRNAs just started a few years ago, and little was known about its function in honey bees, our study should provide important resources for studying lncRNAs with regard to the behavioural plasticity of honey bees.
Methods
Honey bee collections
Honey bees (Apis mellifera) were kept according to standard beekeeping practices at Anhui Agricultural University, Hefei, China. Nurses were collected when they were feeding the larvae inside cells. Foragers were collected when they were flying back to hive with pollen on their hind legs. Four typical colonies were used to provide regularly aged nurses (TC_N) and foragers (TC_F), with N = 30 for each group. One-day-old honey bees from the above four typical colonies were used to create four corresponding single-cohort colonies according to Liu et al. [57] and Ben-Shahar et al. [32]. In short, approximately 1000 one-day-old honey bees were kept in a small hive, which included an unrelated mated queen, an empty comb for the queen to lay eggs, and a comb containing some honey and pollen. In these single-cohort colonies, some honey bees (~ 5–10%) will differentiate into young foragers (7–9 days old, SCC_YF), while others will remain as normally aged (young) nurses (SCC_YN). We then removed the capped brood during the next 30 days, thereby forcing some nurses to continue nursing despite their old age (28–30 days old, SCC_ON), while foragers now were of similar ages (28–30 days old, SCC_OF) to those from typical colonies. We thus decoupled the behaviours of honey bee workers from their ages, which are linked in typical colonies. We sampled 30 bees each from these 4 types of bees from each of the 4 SCCs. All collected honey bees were immediately stored in liquid nitrogen for future RNA extraction.
Library preparation for lncRNA sequencing
Total RNA was isolated from heads of each honey bee sample using TRIzol reagent (Invitrogen, USA). The quality and quantity of the RNA was assessed by the RNA Nano 6000 Assay Kit of the Bioanalyser 2100 system (Agilent Technologies, CA, USA). Three μg of RNA per honey bee sample was used as input material for RNA sample preparations. After removing the ribosomal RNA and rRNA-free residue, we used rRNA-depleted RNA to construct sequencing libraries using a NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (NEB, USA). M-MuLV Reverse Transcriptase, DNA Polymerase I and RNase H were used to obtained the first and the second strand cDNAs respectively. NEBNext Adaptor with a hairpin loop was ligated for hybridization. Finally, the products were purified using AMPure XP system (Beckman Coulter, Beverly, USA), and library quality was assessed with the Agilent Bioanalyser 2100 system. Our libraries were sequenced on an Illumina Hiseq 2500 platform.
Data analysis
Clean reads were obtained by removing reads containing an adapter, reads containing ploy-N and low quality reads from raw data. QC calculation (Q20, Q30 and GC) were performed at the same time. Then clean reads with high quality were used for next analyses. Firstly, all clean reads were mapped to the reference genome (https://www.ncbi.nlm.nih.gov/assembly/GCF_000002195.4/) (http://biomirror.aarnet.edu.au/biomirror/ncbigenomes/Apis_mellifera/GFF//ref_Amel_4.5_top_level.gff3.gz). The index of honey bee genome (Amel 4.5) was built and paired-end clean reads were aligned to the genome. All mapped reads were assembled by both Scripture (beta2) [26] and Cufflinks (v2.1.1) [27]. Cufflinks included the assembler along with the utilities to structurally compare Cufflinks output between samples (Cuffcompare) and to perform differential expression testing (Cuffdiff). Cuffdiff was used to calculate fragments per kilo-base of exon per million fragments mapped (FPKMs) based on the length of the fragments, and the read counts mapped to this fragment for both lncRNAs and coding genes in each sample. Gene FPKMs were computed by summing the FPKMs of transcripts in each gene group. Cuffdiff provides statistical routines for determining differential expression in digital transcript or gene expression data using a model based on the negative binomial distribution. P-adjust< 0.05 and the absolute value of log2 (fold change) > 1 were set as the thresholds for significantly differentially expressed genes.
Screening for provisional lncRNAs
The screening included basic filtering and coding potential filtering (Fig. 6). There were five steps of basic filtering: 1. recurrence: transcripts which were assembled by more than two software or detected in more than two samples were selected; 2. transcript length: transcripts with length of more than 200 bp were selected; 3. minimal read coverage: transcripts with FPKM of more than 2 were selected; 4. filtration of known non-lncRNAs; 5. classification of candidate lncRNAs: Coding Potential Calculator (CPC) and Pfam Scan (Pfam) were used to predict the coding potential. CPC (0.9-r2) was used to assess the protein-coding potential of a transcript based on biologically meaningful sequence features [58]. Pfam (v1.3) was used to identify the occurrence of any of the known protein family domains documented in the Pfam database [59]. Any transcript was excluded if it was predicted to possess coding potential by each of the two methods.

The main workflow for screening lncRNAs
Target gene prediction
In order to study the function of lncRNAs, we used cis and trans to predict the potential targets of lncRNAs. The Cis role is the lncRNA acting on its neighbour genes. We searched protein-coding genes 10 k/100 k upstream and downstream of lncRNAs and then analysed their function. The trans role of lncRNAs was examined based on its expression correlation coefficient with protein coding genes (absolute value of Pearson correlation ≥ 0.95).
GO and KEGG enrichment analyses
Gene Ontology (GO) enrichment analysis of differentially expressed mRNAs or target genes of lncRNAs was performed using the GOseq R package. Kyoto Encyclopedia of Genes and Genomes (KEGG) is a database resource for understanding high-level functions and effects of the biological system (http://www.genome.jp/kegg/). KOBAS (v2.0) was used to test the statistical enrichment of differential expression genes or target genes of lncRNAs in KEGG pathways [58].
Quantitative PCR analysis
Total RNAs from the heads of honey bee from four typical and four single-cohort colonies were used for quantitative PCR analysis. Briefly, the first strand cDNA was obtained using a HiScript II Q RT SuperMix for qPCR (Vazyme, China) and were subjected to quantification of lncRNAs or mRNAs with β-actin as the housekeeping gene using a standard SYBR Green PCR kit (ChamQ™ SYBR Colour Qpcr Master Mix) (Vazyme, China) on the CFX Connect Real-Time System (Bio-Rad). The quantitative PCR was performed under the following conditions: 95 °C for 30 s, 40 cycles of 95.0 °C for 10 s and 60 °C for 30 s. Then, for melting curve analysis, temperatures were increased from 70 °C to 95 °C (at 0.5 °C increment every 5 s until plate reading). Each sample test was performed in triplicate for all reactions. Gene expression was quantified relative to the expression of β-actin using the comparative cycle threshold (ΔCT) method.
Statistical analysis
All qPCR data were analysed using one-way analysis of variance (ANOVA) to test homogeneity of variances via Levene’s test and followed with Students’ T test (PASW Statistics 18.0 software). The results are shown as the mean ± standard error. P < 0.05 was considered to be statistically significant.
Abbreviations
- ACS:
-
Acyl-CoA synthetases
- ACSF2:
-
acyl-CoA synthetase family member 2
- cGMP:
-
cyclic guanosine monophosphate
- CPC:
-
Coding Potential Calculator
- DAMB:
-
dopamine receptor in mushroom bodies
- DNTPs:
-
deoxy-ribonucleotide triphosphate
- dop1:
-
dopamine receptor, D1
- dTTP:
-
deoxy- thymidine triphosphate
- dUTP:
-
deoxyuridine triphosphate
- HR38:
-
hormone receptor-like in 38
- JH:
-
juvenile hormone
- Kr-h1 :
-
Krüppel-homolog 1
- mrjps:
-
major royal jelly proteins
- PFAM:
-
Pfam-scan
- Pkc:
-
protein kinase C
- qPCR:
-
quantitative PCR
- Vg:
-
vitellogenin
References
Robinson GE. Regulation of division of labor in insect societies. Annu Rev Entomol. 1992;37:637–65.
Robinson GE, Page RE, Strambi C, Strambi A. Hormonal and genetic control of behavioral integration in honey bee colonies. Science. 1989;246:109–12.
Huang ZY, Robinson GE. Regulation of honey bee division of labor by colony age demography. Behav Ecol Sociobiol. 1996;39:147–58.
Elekonich MM, Schulz DJ, Bloch G, Robinson GE. Juvenile hormone levels in honey bee (Apis mellifera L.) foragers: foraging experience and diurnal variation. J Insect Physiol. 2001;47:1119–25.
Huang ZY, Robinson GE, Borst DW. Physiological correlates of division of labor among similarly aged honey bees. J Comp Physiol A. 1994;174:731–9.
Robinson GE. Genomics and integrative analyses of division of labor in honeybee colonies. Am Nat. 2002;160:S160–72.
Bloch G, Toma DP, Robinson GE. Behavioral rhythmicity, age, division of labor and period expression in the honey bee brain. J Biol Rhythm. 2001;16:444–56.
The Honeybee Genome Sequencing C. Insights into social insects from the genome of the honeybee Apis mellifera. Nature. 2006;443:931–49.
Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10:155–9.
Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2013;15:7–21.
Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL, Byron M, Monks B, Henry-Bezy M, Lawrence JB, et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science. 2013;341:789–92.
Ding X, Wang X, Lin M, Xing Y, Ge S, Jia R, Zhang H, Fan X, Li J. PAUPAR lncRNA suppresses tumourigenesis by H3K4 demethylation in uveal melanoma. FEBS Lett. 2016;590:1729–38.
Chiu HS, Somvanshi S, Patel E, Chen T-W, Singh VP, Zorman B, Patil SL, Pan Y, Chatterjee SS. Cancer genome atlas research N et al. Pan-Cancer analysis of lncRNA regulation supports their targeting of cancer genes in each tumor context. Cell Rep. 2018;23:297–312.
Maenner S, Müller M, Fröhlich J, Langer D, Becker PB. ATP-dependent roX RNA remodeling by the helicase maleless enables specific association of MSL proteins. Mol Cell. 2013;51:174–84.
Ilik IA, Quinn JJ, Georgiev P, Tavares-Cadete F, Maticzka D, Toscano S, Wan Y, Spitale RC, Luscombe N, Backofen R, et al. Tandem stem-loops in roX RNAs act together to mediate X chromosome dosage compensation in Drosophila. Mol Cell. 2013;51:156–73.
Li M, Wen S, Guo X, Bai B, Gong Z, Liu X, Wang Y, Zhou Y, Chen X, Liu L, et al. The novel long non-coding RNA CRG regulates Drosophila locomotor behavior. Nucleic Acids Res. 2012;40:11714–27.
Yang D, Lian T, Tu J, Gaur U, Mao X, Fan X, Li D, Li Y, Yang M. LncRNA mediated regulation of aging pathways in Drosophila melanogaster during dietary restriction. Aging. 2016;8:2182–97.
Hart RP, Goff LA. Long noncoding RNAs: central to nervous system development. Int J Dev Neurosci. 2016;55:109–16.
Sawata M, Yoshino D, Takeuchi H, Kamikouchi A, Ohashi K, Kubo T. Identification and punctate nuclear localization of a novel noncoding RNA, Ks-1, from the honeybee brain. Rna. 2002;8:772–85.
Sawata M, Takeuchi H, Kubo T. Identification and analysis of the minimal promoter activity of a novel noncoding nuclear RNA gene, AncR-1, from the honeybee (Apis mellifera L.). RNA. 2004;10:1047–58.
Kiya T, Ugajin A, Kunieda T, Kubo T. Identification of kakusei, a nuclear non-coding RNA, as an immediate early gene from the honeybee, and its application for neuroethological study. Int J Mol Sci. 2012;13:15496–509.
Tadano H, Yamazaki Y, Takeuchi H, Kubo T. Age- and division-of-labour-dependent differential expression of a novel non-coding RNA, Nb-1, in the brain of worker honeybees, Apis mellifera L. Insect Mol Biol. 2009;18:715–26.
Jayakodi M, Jung JW, Park D, Ahn YJ, Lee SC, Shin SY, Shin C, Yang TJ, Kwon HW. Genome-wide characterization of long intergenic non-coding RNAs (lincRNAs) provides new insight into viral diseases in honey bees Apis cerana and Apis mellifera. BMC Genomics. 2015;16:680.
Chen X, Ma C, Chen C, Lu Q, Shi W, Liu Z, Wang H, Guo H. Integration of lncRNA-miRNA-mRNA reveals novel insights into oviposition regulation in honey bees. PeerJ. 2017;5:e3881.
Wedd L, Kucharski R, Maleszka R. Differentially methylated obligatory epialleles modulate context-dependent LAM gene expression in the honeybee Apis mellifera. Epigenetics. 2016;11:1–10.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5.
Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, et al. Ab initio reconstruction of cell type–specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol. 2010;28:503–10.
Legeai F, Derrien T. Identification of long non-coding RNAs in insects genomes. Curr Opin Insect Sci. 2015;7:37–44.
Soshnev AA, Ishimoto H, McAllister BF, Li X, Wehling MD, Kitamoto T, Geyer PK. A conserved long noncoding RNA affects sleep behavior in Drosophila. Genetics. 201; 189: 455.
Chen B, Zhang Y, Zhang X, Jia S, Chen S, Kang L. Genome-wide identification and developmental expression profiling of long noncoding RNAs during Drosophila metamorphosis. Sci Rep. 2016;6:23330.
Satyavathi V, Ghosh R, Subramanian S. Long non-coding RNAs regulating immunity in insects. Non-Coding RNA. 2017;3:14.
Ben-Shahar Y, Robichon A, Sokolowski MB, Robinson GE. Influence of gene action across different time scales on behavior. Science. 2002;296:741–4.
Watkins PA. Fatty acid activation. Prog Lipid Res. 1997;36:55–83.
Sivachenko A, Gordon HB, Kimball SS, Gavin EJ, Bonkowsky JL, Letsou A. Neurodegeneration in a Drosophila model of adrenoleukodystrophy: the roles of the bubblegum and double bubble acyl-CoA synthetases. Dis Model Mech. 2016;9:377–87.
Toth AL, Robinson GE. Worker nutrition and division of labour in honeybees. Anim Behav. 2005;69:427–35.
Amdam GV, Norberg K, Hagen A, Omholt SW. Social exploitation of vitellogenin. PANS. 2003;100:1799–802.
Ihle KE, Page RE, Frederick K, Fondrk MK, Amdam GV. Genotype effect on regulation of behaviour by vitellogenin supports reproductive origin of honeybee foraging bias. Anim Behav. 2010;79:1001–6.
Drapeau MD, Albert S, Kucharski R, Prusko C, Maleszka R. Evolution of the yellow/major Royal Jelly Protein family and the emergence of social behavior in honey bees. Genome Res. 2006;16:1385–94.
Malecova B, Ramser J, O'Brien JK, Janitz M, Judova J, Lehrach H, Simuth J. Honeybee (Apis mellifera L.) mrjp gene family: computational analysis of putative promoters and genomic structure of mrjp1, the gene coding for the most abundant protein of larval food. Gene. 2003;303:165–75.
Hojo M, Kagami T, Sasaki T, Nakamura J, Sasaki M. Reduced expression of major royal jelly protein 1gene in the mushroom bodies of worker honeybees with reduced learning ability. Apidologie. 2010;41:194–202.
Whitfield CW, Cziko AM, Robinson GE. Gene expression profiles in the brain predict behavior in individual honey bees. Science. 2003;302:296–9.
Ben-Shahar Y, Dudek NL, Robinson GE. Phenotypic deconstruction reveals involvement of manganese transporter malvolio in honey bee division of labor. J ExpBiol. 2004;207:3281–8.
Blenau W, Erber J, Baumann A. Characterization of a dopamine D1 receptor from Apis mellifera: cloning, functional expression, pharmacology, and mRNA localization in the brain. J Neurochem. 1998;70:15–23.
Dis. Model. Mech. DAMB, a novel dopamine receptor expressed specifically in Drosophila mushroom bodies. Neuron. 1996;16:1127–35.
Dong D, Zhang Y, Smykal V, Ling L, Raikhel AS. HR38, an ortholog of NR4A family nuclear receptors, mediates 20-hydroxyecdysone regulation of carbohydrate metabolism during mosquito reproduction. Insect Biochem Mol Biol. 2018;96:19–26.
Khamis AM, Hamilton AR, Medvedeva YA, Alam T, Alam I, Essack M, et al. Insights into the transcriptional architecture of behavioral plasticity in the honey bee Apis mellifera. Sci Rep. 2015;5:11136.
Yamazaki Y, Shirai K, Paul RK, Fujiyuki T, Wakamoto A, Takeuchi H, Kubo T. Differential expression of HR38 in the mushroom bodies of the honeybee brain depends on the caste and division of labor. FEBS Lett. 2006;580:2667–70.
Kusserow A, Pang K, Sturm C, Hrouda M, Lentfer J, Schmidt HA, et al. Unexpected complexity of the Wnt gene family in a sea anemone. Nature. 2005;433:156–60.
Dearden PK, Wilson MJ, Sablan L, Osborne PW, Havler M, McNaughton E, et al. Patterns of conservation and change in honey bee developmental genes. Genome Res. 2006;16:1376–84.
Abdou M, Peng C, Huang J, Zyaan O, Wang S, Li S, Wang J. Wnt signaling cross-talks with JH signaling by suppressing Met and gce expression. PLoS One. 2011;6:e26772.
Robinson GE. Regulation of honey bee age polyethism by juvenile hormone. Behav Ecol Sociobiol. 1987;20:329–38.
Sullivan JP, Fahrbach SE, Robinson GE. Juvenile hormone paces behavioral development in the adult worker honey bee. Horm Behav. 2000;37:1–14.
Oliva CA, Montecinos-Oliva C, Inestrosa NC. Wnt signaling in the central nervous system: new insights in health and disease. In: Larraín J, Olivares G, editors. Progress in molecular biology and translational science: Academic Press; 2018. p. 81–130.
Maguschak KA, Ressler KJ. A role for WNT/beta-catenin signaling in the neural mechanisms of behavior. J Neuroimmune Pharm. 2012;7:763–73.
Humphries MA, Muller U, Fondrk MK, Page REJ. PKA and PKC content in the honey bee central brain differs in genotypic strains with distinct foraging behavior. J Comp Physiol A. 2003;(7):555–62.
Kane NS, Robichon A, Dickinson JA, Greenspan RJ. Learning without performance in PKC-deficient Drosophila. Neuron. 1997;18:307–14.
Liu F, Shi T, Yin W, Su X, Qi L, Huang ZY, Zhang S, Yu L. The microRNA ame-miR-279a regulates sucrose responsiveness of forager honey bees (Apis mellifera). Insect Biochem Mol Biol. 2017;90:34–42.
Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei L, Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35:W345–9.
Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–301.
Acknowledgements
We thank for TD Liang for assistance with bees and RNA extraction; XL Liu and SZ Huang for technical assistance in the field; K Zahrah for reviewing the manuscript.
Funding
This work was supported by National Natural Science Foundation of China (Grant no. 31302039) and University Natural Science Research Project of Anhui Province (Grant no. KJ2018A0138). The funding bodies had no role in the design of the study and collection, analysis and interpretation of data and in writing the manuscript.
Availability of data and materials
The sequencing data are available in the SRA database (SRP153124) of the NCBI system.
Author information
Authors and Affiliations
Contributions
FL conceived the idea for this research, performed data analysis, and wrote the manuscript. TFS performed RNA extraction, RT-PCR and qPCR analysis. LQ, XS and JD participated in data analysis and honey bee sampling. DQW provided the experimental environment and coordination. ZYH performed the statistical analysis and wrote the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Detailed information on six honey bee samples for RNA-seq. (XLS 25 kb)
Additional file 2:
The distribution of reads mapped to reference annotation. (XLS 25 kb)
Additional file 3:
A hierarchical heat map showing the transformed expression values for transcripts (mRNA and lncRNA). Red shows higher expression, and blue shows lower expression. (PPTX 347 kb)
Additional file 4:
Common differentially expressed lncRNAs and mRNAs among the three comparisons. (XLS 134 kb)
Additional file 5:
Venn diagram of common differential expression transcripts (lncRNA and mRNA) among two comparison groups (SCC_YF vs. SCC_YN and SCC_OF vs. SCC_ON). (PPTX 175 kb)
Additional file 6:
The top 20 enriched pathways in the TC_F vs. TC_N (A), SCC_YF vs. SCC_YN (B) and SCC_OF vs. SCC_ ON (C) comparisons (cis). (XLSX 12 kb)
Additional file 7:
The significantly enriched GO terms (corrected p-value < 0.05) detected in the TC_F vs. TC_N, SCC_YF vs. SCC_YN and SCC_OF vs. SCC_ON comparisons (trans). (XLS 68 kb)
Additional file 8:
The top 20 enriched KEGG pathways in the TC_F vs. TC_N, SCC_YF vs. SCC_YN and SCC_OF vs. SCC_ ON comparisons (trans). (XLS 30 kb)
Additional file 9:
The significantly enriched GO terms (corrected p-value < 0.05) of differentially expressed mRNAs from the three comparison groups. (XLS 74 kb)
Additional file 10:
The top 20 enriched KEGG pathways of differentially expressed mRNAs from the three comparison groups. (XLS 31 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liu, F., Shi, T., Qi, L. et al. lncRNA profile of Apis mellifera and its possible role in behavioural transition from nurses to foragers. BMC Genomics 20, 393 (2019). https://doi.org/10.1186/s12864-019-5664-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-5664-7