
Abstract
Background
Long noncoding RNAs (LncRNAs) play important roles in fundamental biological processes. However, knowledge about the genome-wide distribution and stress-related expression of lncRNAs in tilapia is still limited.
Results
Genome-wide identification of lncRNAs in the tilapia genome was carried out in this study using bioinformatics tools. 103 RNAseq datasets that generated in our laboratory or collected from NCBI database were analyzed. In total, 72,276 high-confidence lncRNAs were identified. The averaged positive correlation coefficient (r_mean = 0.286) between overlapped lncRNA and mRNA pairs showed significant differences with the values for all lncRNA-mRNA pairs (r_mean = 0.176, z statistics = − 2.45, p value = 0.00071) and mRNA-mRNA pairs (r_mean = 0.186, z statistics = − 2.23, p value = 0.0129). Weighted correlation network analysis of the lncRNA and mRNA datasets from 12 tissues identified 21 modules and many interesting mRNA genes that clustered with lncRNAs. Overrepresentation test indicated that these mRNAs enriched in many biological processes, such as meiosis (p = 0.00164), DNA replication (p = 0.00246), metabolic process (p = 0.000838) and in molecular function, e.g., helicase activity (p = 0.000102) and catalytic activity (p = 0.0000612). Differential expression (DE) analysis identified 99 stress-related lncRNA genes and 1955 tissue-specific DE lncRNA genes. MiRNA-lncRNA interaction analysis detected 72,267 lncRNAs containing motifs with sequence complementary to 458 miRNAs.
Conclusions
This study provides an invaluable resource for further studies on molecular bases of lncRNAs in tilapia genomes. Further function analysis of the lncRNAs will help to elucidate their roles in regulating stress-related adaptation in tilapia.
Similar content being viewed by others

Background
Long non-coding RNAs (lncRNAs) are transcripts longer than 200 nt but without coding potential. They play roles in transcriptional regulation and post-transcriptional regulation, and show functions in protein localization, telomere replication, and RNA interference [1,2,3,4] and conservation in Vertebrates [5]. With the development of whole genome RNA sequencing (RNA-Seq) technique and computational analysis, genome-wide identification of lncRNAs have been performed in many species, such as yeast [6], fruit fly [7], Atlantic salmon [8], zebrafish [9], chicken [10], rat [11] and human [12].
LncRNAs were closely associated with stress responses in animals. Ambient stressors, e.g., chemistry treatment [13,14,15], drug reaction [16], high salt [17] and pathogen infection [8] were found to induce lncRNA expression changes. For example, in mice, lncRNA TUG1 exerted a protect effect against cold-induced liver damage by inhibiting apoptosis [18].
A lot of studies have paid attention to characterize the transcriptional responses to cold stress in fishes such as common carp (Cyprinus carpio) [19], zebrafish (Danio rerio) [20,21,22], channel catfish (Ictalurus punctatus) [22], killifish (Austrofundulus limnaeus) [23], coral reef fish (Pomacentrus moluccensis) [24] and rainbow trout (Oncorhynchus mykiss) [25]. Gene expression changes in response to hypoxia were explored in some teleosts, such as in divergent tissues of adults of the euryoxic gobiid fish Gillichthys mirabilis [26], the gills [27] and the heart [28] of zebrafish and zebrafish embryos [29], the heart and liver in the teleost fish Fundulus grandis [30] and in Tilapia [31, 32]. However, there was very limited stress-related work on lncRNAs in economic fish. In rainbow trout [33] and Atlantic salmon [8], differential expressions of lncRNAs in response to infection were reported.
Tilapia is one of the most important cultural fish worldwide with great economic importance [34]. In tilapia, much attention has been paid to stress responses. The gene expression changes in response to different stressors including cold stress [35], alkalinity stress [36], salinity adaptation [37], hypoxia stress [31, 32, 38] have been investigated by RNA-seq. The QTL intervals for hypoxia and salt tolerance traits were mapped recently [39, 40]. Function of miRNAs was investigated under high-salt, alkalinity, hypoxia, Streptococcus agalactiae infection and other adverse conditions [41,42,43,44,45,46,47,48]. LncRNAs were relatively new class of RNA molecules that were less well characterized in tilapia. Recently, Hezroni et al. identified lncRNAs based on one tilapia RNAseq dataset and evaluated the conservation of lncRNAs in vertebrates [5]. The expression profile of 797 putative non-coding transcripts in tilapia macrophages activated by HSP70 and Streptococcus agalactiae antigen was reported [49].
In this study, to uncover lncRNA characteristics in tilapia genome, we generated and collected a total of 103 RNA-seq samples and carried out a genome-wide identification of lncRNAs. The potential association of the lncRNAs with different stressors in different tissues was analyzed. Our finding provided an invaluable resource for further studies on molecular basis of lncRNAs in tilapia.
Results
Genome-wide identification of lncRNAs in tilapia
Understanding the characteristics of lncRNAs in tilapia genome would be useful for exploring functional mechanisms related to economic traits. To obtain a full annotation of lncRNAs in tilapia genome, we totally generated 32 RNAseq datasets in our laboratory and collected 71 RNAseq datasets from NCBI SRA database (Additional file 1: Table S1). These samples originated from at least 15 different tissues and different stress conditions, e.g. hypoxia, salinity, Fadrozole, lipid content, alkalinity, temperature, Streptococcus agalactiae infection or show differences in other economic traits, such as body colors. This dataset represents the largest data collection for identification of lncRNAs in tilapia.
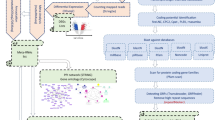
A step-wise protocol was applied to ensure a high-confident discovery of lncRNAs (Fig. 1). The libraries were first mapped to the reference tilapia genome (Orenile1.0) and then the successfully mapped high quality reads were used for transcriptome assembly. The de novo assembly of the 103 RNAseq data produced 321,154 transcripts with a length more than 300 bp.

The flow chart for identification of lncRNAs in the study
To validate the assembly, all transcripts were searched against 10 tilapia Transcriptome Shotgun Assembly (TSA) databases by using Blastn (Additional file 1: Table S1). The TSA datasets contained 1,189,903 sequences with a minimum length of 200 bp to a maximum length of 27,441 bp. The average sequence length was 1111 bp. The search indicated 186,333 transcripts in our assembly showing high similarity and an average coverage of 58% to 105,960 transcripts in the TSA datasets. The remaining transcripts with no significant hits in the TSA databases suggested lots of novel transcripts found in our dataset.
From the assembly, 102,284 lncRNA candidates were initially predicted. Further coding potential analysis suggested that 79,017 of the transcripts were candidate lncRNAs. The candidates near gaps in the genome might represent truncated coding genes. We further filtered the lncRNA candidates that located within 100 bp of the downstream and upstream of gaps. The remaining 72,276 transcripts were denoted as lncRNAs in this study (Table 1).
The lengths for these transcripts ranged from 301 bp to 7783 bp with an average sequence length of 764 bp, a N50 of 832 bp and total bases of 55,238,101 (Table 1 and Additional file 2: Figure S1). The exon numbers for lncRNAs ranged from one to eight exons with an average of 1.1. Most of the lncRNAs (~ 94%) were single-exon types. The average exon number for all transcripts in the de novo assembly was 8.9 with a range from 1 to 219 exons.
Microsatellite instability is a sign of DNA mismatch repair deficiency that can be inherited, which is involved in regulation of lncRNA gene function, e.g. CCAT2 in colorectal cancer [50]. We observed 4788 lncRNAs contained 1-10 bp microsatellite repeat units. 3638 of the microsatellites were classified to the category of 2-bp repeat units (Table 1). This data will be helpful for exploring the function of microsatellite-contained lncRNAs in fish.
Identifications of the lncRNAs against the predicted 6455 noncoding RNA data (Orenil1.0.ncrna downloaded from Ensemble database) using Blastn, we found that 3263 Ensemble noncoding RNAs showed high similarity to 2532 lncRNAs in this study (Identity > 90%, <E-6) with a mean coverage length of 709 bp. This suggests a lot of noncoding RNAs have been captured in our data.
Co-expression analysis between lncRNAs and mRNAs
LncRNAs were generally co-expressed with their neighboring genes [51]. To investigate the relationship between lncRNAs and mRNA genes in tilapia genome, we first identified mRNA candidates from the uncertain transcripts in the assembly by searching against the mRNA database (GCF_001858045.1_ASM185804v2_rna) and protein database (Oreochromis_niloticus.Orenil1.0.pep.all.fa; ID> 99%, E0). Finally 42,634 transcripts were considered to be mRNA candidates for further analysis. This may be lowly representative of the mRNAs in our dataset.
The position data of lncRNAs (4596) and mRNA candidates (42,634) with strand information that generated in the gtf file by Cuffmerge were retrieved. The gene expression data for all samples were used for correlation analysis. The average gene expression value across the 103 samples was 8322 normalized read counts for mRNAs and 1800 normalized read counts for lncRNAs. These suggest the lncRNAs were much lower expressed compared to the mRNAs, similar to previous reported such as in cashmere goat [52].
The mean negative correlation coefficient (r_mean) was ranging from − 0.049 to − 0.069 for all pairs and showed no significant differences (Fig. 2). We observed that the mean positive correlation coefficient (r_mean) was 0.286 for 444 lncRNA-mRNA pairs that overlapped with at least one basepair. This r_mean value was generally larger than the values for non-overlapped lncRNA-mRNA data (e.g., from 0.281 to 0.250 for lncRNA-mRNA pairs within 1-500 bp to 5500-6000 bp distances) and showed significant differences with the values for all lncRNA-mRNA pairs (r_mean = 0.176, z statistics = − 2.45, p value = 0.00071) and all mRNA-mRNA pairs (r_mean = 0.186, z statistics = − 2.23, p value = 0.0129) (Fig. 2).

The relationship between positive and negative correlation coefficients (r_mean) and lncRNA-mRNA distances. The positive and negative r_mean for all lncRNAs, mRNAs and lncRNA-mRNA pairs were plotted. ‘*’ and ‘**’, indicate significant differences (p < 0.05 or p < 0.001) of positive r_mean values compared to the r_mean of overlapped lncRNA-mRNA pairs as assessed using the Fisher r-to-z transformation statistics
To identify functional gene modules across different tissues, we randomly selected 36 RNAseq samples that originated from 12 different tissues (three samples per tissue). The lncRNA-mRNA pairs with high correlation coefficient (|r| > 0.7) including 3844 mRNAs and 1255 lncRNAs were kept for weighted Gene Co-expression network analysis (WGCNA). The cluster analysis of the lncRNA expression indicated that most samples were grouped according to the origins of tissues (Additional file 3: Figure S2).
The WGCNA analysis identified 21 modules with a transcript number from 35 to 1244 in each module. The network heatmap plot for the transcripts was present in Fig. 3. The lncRNA-mRNA pairs with the Topological Overlap Matrix (TOM) similarity values > 0.5 were presented in Additional file 4: Table S2. An example network for ‘grey60’ module was shown in Additional file 5: Figure S3.

Visualizing the gene network for RNAseq dataset using a heatmap plot. The heatmap depicts the Topological Overlap Matrix (TOM) among all genes in the analysis. Light color represents low overlap and progressively darker red color represents higher overlap. Blocks of darker colors along the diagonal are the modules. The gene dendrogram with dissimilarity based on topological overlap and module assignment are also shown along the left side and the top
Overrepresentation test indicated that these mRNAs were enriched in a few biological processes, such as meiosis (p = 0.00164), DNA replication (p = 0.00246), metabolic process (p = 0.000838) and in molecular function, e.g., helicase activity (p = 0.000102) and catalytic activity (p = 0.0000612). The detail information of the overrepresentation test on GO-slim classification was put in Additional file 6: Table S3. The dataset may provide a basis for exploration of the lncRNA function in tilapia.
Identification of stress-related lncRNAs
To characterize the stress-related lncRNAs in tilapia, the collected samples without replicates and stress challenges were firstly filtered. We finally kept 12 RNAseq samples in response to 24 h’s cold challenge that generated in this study and collected two published datasets under hypoxia and salinity challenges. The clean reads were first mapped to the de novo transcriptome assembly. The gene expression profiles were used in DE analysis. Twenty DE transcripts were randomly selected for validation by qRT PCR. PCR primers were successfully developed for 15 of these genes (Additional file 7: Table S4). Except the two transcripts (TCONS_00094910 and TCONS_00010892), the remaining 13 transcripts that revealed by qRT PCR showed same regulation directions as the RNAseq data (r = 0.73). Therefore, the qRT PCR experiments indicated a well concordance with the RNAseq expression data.
In brain under 24 h’s cold treatment, 42 DE lncRNA genes (FDR < 0.05) including 31 down-regulated and 11 up-regulated genes were identified. In heart under 24 h’s cold treatment, 28 DE lncRNA genes were observed including 6 down-regulated and 22 up-regulated genes. In gill under hypoxia challenge, 19 DE lncRNA genes (FDR < 0.05) including 13 down-regulated and 6 up-regulated were identified. In heart under hypoxia challenge, 14 DE lncRNA genes (FDR < 0.05) including 6 down-regulated and 8 up-regulated were identified. Only 2 DE lncRNA genes were found in intestines under seawater challenges. The summary statistics was put in Table 2 and the detail information was presented in the Additional file 8: Table S5.
Of the 99 unique DE lncRNA genes, only 6 simultaneously showed significant responses to two stress conditions. The lncRNA lnc_TCONS_00002141 were significantly differentially expressed in heart under cold and hypoxia treatment. lnc_TCONS_00214756 and lnc_TCONS_00290917 were significantly unregulated, but lnc_TCONS_00047189, lnc_TCONS_00226316 and lnc_TCONS_00151992 were significantly down-regulated in brain and heart under cold treatment.
We found that 5 DE lncRNA genes (lnc_TCONS_00082453, lnc_TCONS_00105572, llnc_TCONS_00144691, llnc_TCONS_00186275 and lnc_TCONS_00290917) were co-expressed with 331 mRNA genes as revealed by WGCNA network analysis. 33 of these genes (e.g., Acads, Adsl, Bard1, Calcoco2, Ccna2, Ccnb1, Cdc7, Cdk7, Cul3, Cxcr4, Dab2, Dapk1, Eif2b3, Eme1, Exo1, Fancg, Fntb, G6pd, Hnmt,Ldha, Lig4, Morf4l1, Msh2, Nfkbil1, P2rx7, Pif1, Pold3, Ppp3ca, Tbl2, Tmem8c, Tsc22d2, Ube4a, Vldlr) were mapped to the GO term GO:0006950 (response to stress).
Tissue-specific DE lncRNA genes
We identified 1955 DE lncRNA genes with 1083 up-regulated and 871 down-regulated lncRNAs for all tested tissues. Except the DE lncRNA genes that identified in brain, heart and ovary, in which most of the lncRNAs were down-regulated, the DE lncRNA genes were absolutely up-regulated in remaining tissues, such as in spleen, skin and liver (Table 2 and Additional file 9: Table S6). We also observed many lncRNAs were shared by at least two tissues (Additional file 10: Figure S4). The distribution features was similar to previous studies that lncRNAs are frequently expressed in a tissue-specific fashion [53] and 90% of the lncRNAs are shared by at least two tissues in fish [8]. These findings indicated that the lncRNAs may regulate biological processes through their spatio-temporal expression.
Interaction between lncRNAs and miRNAs
It is becoming increasingly evident that regulation of gene expression through lncRNAs as competing endogenous RNAs, such as competition with miRNA binding, is a general phenomenon. To characterize the interaction of miRNAs and lncRNAs, we carried out a microRNA-lncRNA interaction analysis with miRanda (ver.3) [54]. Higher alignment procedure scores based on sequence complementarity and lower minimum free energy (MFE) values reveal better thermodynamic stability of RNA duplexes [55]. We found 72,267 lncRNAs contain a motif with sequence complementary to 458 miRNAs under a strict filter parameters (miRanda total score > 120, minimum free energy <− 12 kcal/mol and matched length > =18 bp). The average score is 148.1 with a maximum score of 205. The average minimum free energy is − 17.92 (kcal/mol) with the lowest free energy of − 49.3 (kcal/mol). The 8878 highly reliable miRNA-lncRNA interaction pairs with a low free energy from − 30 to − 49.3 (kcal/mol) were provided in Additional file 11: Table S7.
The 99 stress-related DE lncRNA genes contained a motif with sequence complementary to 448 miRNAs. Ten of them contained a motif with sequence complementary to 17 mature miRNAs and a free energy less than − 30 kcal/mol. The pair of lncRNA ‘TCONS_00151992’ and miRNA ‘dre-miR-128-5p’ have the lowest minimum free energy (− 36.69 kcal/mol) and a maximum score of 175. These data suggest these miRNA molecules may involve into stress responses.
Discussions
Large data size of lncRNAs found in tilapia
The average sequence length of lncRNAs identified in this study was much shorter than that of the NCBI reference RNA data (an average length of 3216 bp and a N50 of 4383 bp; GCF_001858045.1_ASM185804v2_rna.fa) for tilapia, suggesting lncRNAs had shorter transcripts than protein-coding genes. This feature was generally discovered in lncRNA identification, such as in rice brown planthopper (Nilaparvata lugens) [56] and silkworm [57]. Besides, the exon number for lncRNAs(1.1) was much less than other types of genes(8.9)in tilapia. The finding was consistent to the previous reports, such as in Luo et al. [49] that the predicted lncRNAs contained fewer exons than protein genes. Moreover, the differences between the number of identified lncRNAs in this study and Ensemble noncoding RNAs can be due to the differences in the pipelines applied for their identification.
The human genome might harbor nearly as many lncRNAs as protein-coding genes (perhaps 15,000 lncRNA), although only a fraction were expressed in a specific cell type [58]. In the pig genome, 6621 lncRNAs were identified by analyzing 93 samples and expressed sequence datasets [59]. In Anopheles gambiae [60] and Drosophila melanogaster [61,62,63] around 3000 lncRNA genes were identified. In fish, 21,065 high-confident non-coding transcripts were discovered in Salmo salar [8] and 16,600 to 33,600 unique lncRNAs in zebrafish were found [16]. The lncRNA data size identified in the tilapia genome was more than the reported data, partially because of the large samples from different conditions and tissues used in the study.
A few co-expression profiles for lncRNAs and mRNAs worth further investigation
The prediction of lncRNAs and mRNA interactions is very important to study the function of lncRNAs. The genes on the same pathways or in the same functional complex often exhibit similar expression patterns under diverse temporal and physiological conditions [64]. The genes show a similar expression pattern across samples and tissues can be inferred by analyzing their co-expression networks [65, 66]. The gene co-expression networks have become a rapidly developing area of study and many interesting results have been obtained, such as genes related to mouse weight [67].
In tilapia, the averaged positive correlation coefficient (r_mean = 0.286) between overlapped lncRNA and mRNA pairs showed significant differences with the values for all lncRNA-mRNA pairs (r_mean = 0.176, z statistics = − 2.45, p value = 0.00071) and mRNA-mRNA pairs (r_mean = 0.186, z statistics = − 2.23, p value = 0.0129). This suggests lncRNAs may have roles in cis-regulating their neighboring protein coding genes [51]. Moreover, the presence of shared regulatory elements controlling the expression of both promoters can be another possible interpretation.
We also found 55,121 pairs of lncRNA and mRNA transcripts with a TOM value range of 0.5 to 0.93. These transcripts contained 462 lncRNAs and 1023 mRNAs. A few known co-expression profiles have been found in the dataset. For example, lnc_TCONS_00284667 shows similar co-expression profiles with gene dmrt1 (TOM = 0.52) and tex11 (TOM = 0.52). Dmrt1 has been known to be involved in the process of sex determination [68]. Tex11 is associated with meiosis [69]. lnc_TCONS_00271192 is closely associated with gene slc13a1 (TOM = 0.57), slc22a13 (TOM = 0.69), slc22a7 (TOM = 0.62) and slc2a11a (TOM = 0.67). Based on earlier findings, slc13a1, slc22a6 and slc2a11 were co-expressed in human pluripotent stem cell [70]. In addition, co-expression of slc13a1 and slc22a6 was identified in transcriptional profiling of human central nervous system [71]. Hence, our study reveals closely associated relationship among genes and/or lncRNAs. However, there is little information available on the co-expression of many modulated genes. For example, lnc_TCONS_00109777 showed strong co-expression with gene enosf1 (TOM = 0.89), march7 (TOM = 0.93), cep290 (TOM = 0.93) and hps3 (TOM = 0.93). These results may worth further investigation.
lncRNAs might play key roles in response to different stressors in tilapia
Differentially expressed (DE) analysis of the RNAseq datasets have been conducted to identify lncRNAs significantly in response to stress. For example, non-coding transcriptional response induced by pathogens has been characterized in infected salmons [8, 72] and in zebrafish under b-diketone antibiotic exposure [73].
Improvement of the stress tolerance, e.g., low-temperature, hypoxia and salt tolerance has become an important issue for aquaculture development of tilapia. The transcriptional responses to cold stress and hypoxia were explored in some teleosts [19,20,21,22,23,24,25,26,27,28,29,30,31,32]. In tilapia, the gene expression changes in response to alkalinity stress [36], salinity adaptation [37] have been carried out. These investigations have revealed a large number of stress responsible genes involved in a variety of biological processes. LncRNAs play important roles in stress responses. For example, LncRNAs TUG1 and H19 transcript levels were elevated at 1.94-fold and 2.44-fold, respectively, in skeletal muscle of hibernating ground squirrels compared with euthermicones [74]. In mice, lncRNA TUG1 exerted a protect effect against cold-induced liver damage by inhibiting apoptosis [18]. However, little information on the function of lncRNAs in cold, hypoxia and salinity stresses are available in fish. In this study, the identification of lots of differentially expressed lncRNA genes in heart, brain and intestines in response to cold stress, salt or hypoxia stress suggested that lncRNAs might play key roles in response to different stressors in tilapia.
lncRNAs might act in miRNA function
The crosstalk between lncRNAs and miRNAs is intricate and complex. Several studies have indicated that the lncRNAs can enhance pri-miRNA processing or act as precursor [75,76,77]. In this study, we found 72,267 lncRNAs contain a motif with sequence complementary to 458 miRNAs. Of which, 99 stress-related DE lncRNA genes contained a motif with sequence complementary to 448 miRNAs. In addition, only low proportions of miRNA-lncRNAs pairs (~ 2%) show well complementary (100% identity) (Additional file 11: Table S7). This means very few lncRNAs may act as precursors in tilapia. Coding and non-coding RNA transcripts with shared miRNA response elements (MREs) were able to actively communicate with each other [78, 79]. Previously report indicated that when a given mRNA was up-regulated, the repression conferred by its associated targeting miRNAs was decreased, as the total number of MREs exceeds that of the miRNAs themselves [80]. However, the relationship among mRNA, lncRNA and miRNAs is still unclear. Our work provides a valuable non-coding resource to evaluate transcriptional modulation of lncRNAs under stress challenge in future.
Conclusions
A computational pipeline based on 103 RNAseq datasets identified 72,276 lncRNAs from the tilapia genome. Co-expression network analysis identified 21 modules and important clustered genes in different tissues. DE analysis identified 99 DE lncRNA genes in response to cold, salt and hypoxia stressors and 1955 DE lncRNA genes for all tested tissues. This study provided an invaluable resource for further studies on molecular bases of lncRNAs in tilapia genomes. Further function analysis of the lncRNAs is required to elucidate their roles in regulating stress-related adaptation.
Methods
Fish management, cold treatment, sample collection
To identify cold stress-associated lncRNAs in tilapia, we randomly selected 36 Nile tilapia individuals (~ 90 dph) from a population raised at the fish facility of School of Life Science, Sun Yat-Sen University. These fishes were randomly divided into two indoor conical fibre glass tanks (water depth: 70 cm, volume: 500 L) in a recirculating freshwater system for acclimation of 2 days with water temperature at 25–28 °C and dissolved oxygen (DO) > 6 mg L− 1. The fishes were fed with tilapia pellets twice a day and the photoperiod was adjusted to 12D:12 L in the room.
During low temperature treatment, the fishes in one tank were used as the control and those in the other were considered as test samples. The temperature in the water of test tank was slowly decreased to 15 °C within 12 h using a water cooler. The control tank was maintained under 25–28 °C. Following 24 h cold treatment, the fishes were anesthetized with MS 222 before we collected six tissue samples including brain and heart from each fish. The tissue samples were kept immediately in TRIzol reagent (Invitrogen, UK) and then stored at − 80 °C.
RNA isolation and NGS sequencing
Total RNA from cold-treated samples was isolated using TRIzol reagent (Invitrogen, UK) according to the manufacturer’s protocol. The RNA quality was assessed using the Nanodrop-2000 (Thermo Scientific, USA) and electrophoresis in 1.5% agarose Gel. Total RNA integrity was further evaluated by using Bioanalyzer 2100 (Agilent Technologies). Twelve libraries from heart and brain samples that constructed by TruSeq™RNA Sample Preparation Kit according to the product instruction (Illumina) were finally sequenced using Illumina HiSeq2500 for 2 × 150 bp pair-end (PE) sequencing.
LncRNA identification from RNA-seq dataset
To maximize the identification of lncRNAs in tilapia genome, 12 cold–treated samples and 20 hypoxia-treated RNAseq datasets from brain, spleen, heart and gills that previously generated in our laboratory [31, 32] and other 71 RNAseq datasets downloaded from NCBI SRA database from different tilapia tissues were also collected in this study (Additional file 1: Table S1).
The RNA-seq data from each sample was first mapped to the tilapia reference genome (Orenile1.0 and Orenil1.0.88.gtf) by using TopHat [81]. The mapped reads then was de novo assembled using the program Cufflinks and Cuffmerge with default parameters [82]. The fasta sequences were generated using the program Gffread (https://github.com/gpertea/gffread) and the sequences with a length less than 300 bp were filtered. To validate the assembly, sequence similarity search of all the transcripts against 10 tilapia Transcriptome Shotgun Assembly (TSA) databases (Additional file 1: Table S1) with the basic local alignment search tool Blastn [83]. All hits with an E value <1e-6 and identity (ID) > = 98% were considered as significant blast hits.
PLEK [84] uses an improved computational pipeline based on k-mer and support vector machine (SVM) to distinguish long non-coding RNAs (lncRNAs) from messenger RNAs (mRNAs). CNCI [85] by profiling adjoining nucleotide triplets can effectively distinguish protein-coding and non-coding sequences independent of known annotations to retrieve novel lincRNAs. The lncRNAs from our transcriptome assembly were initially predicted using the software PLEK [84] with score < − 0.5 and CNCI [85] with default parameters.
Coding potential analysis of the non-coding sequences were conducted using CPC [86] and TransDecoder-3.0.0 (https://github.com/TransDecoder/TransDecoder/wiki). The candidate lncRNA datasets from the output of CPC were translated into proteins by using the software TransDecoder-3.0.0. LncRNAs contain an ORF longer than 100 amino acids were removed. The longest ORFs for the remaining candidates were then searched against Swissprot and Pfam protein database (release 27; used both Pfam A and Pfam B) (http://pfam.xfam.org/) using the program hmmscan (www.ebi.ac.uk/Tools/hmmer/search/hmmscan) and Blastp [83]. All hits with E values <1e-6 were considered as significant blast hits. The final lncRNA dataset should not have similarity to any known proteins. LncRNA candidates near gaps in the genome might represent truncated coding genes. We further filtered the lncRNA candidates that located within 100 bp of the downstream and upstream of gaps. The remaining transcripts were denoted as lncRNAs in this study (Additional file 12). The lncRNAs candidates were compared with Orenil1.0.ncrna dataset that downloaded from Ensembl genome database (www.ensembl.org).
Genome location, distances between pairs of mRNAs and lncRNAs, and correlation of gene expression profiles
The remaining uncertain transcripts were searched against the tilapia protein database (Oreochromis_niloticus.Orenil1.0.pep.all.fa) by Blastx and mRNA_sequences from dataset GCF_001858045.1_ASM185804v2_rna_by Blastn with parameters ID > = 99% and E-value = 0. The transcripts with significant hits as revealed by both programs were considered to be mRNA candidates. The positions for lncRNAs and mRNA candidates with stranded information were then retrieved from the merged.gtf that generated using Cuffmerge. The distances for each pair of lncRNA and mRNA genes were calculated using shell scripts. The gene counts that normalized with the reads per kilobase per million mapped reads (RPKM) normalization approach were obtained by using the program Cuffnorm (http://cole-trapnell-lab.github.io/cufflinks/cuffnorm/). The correlation coefficients of gene expressions for pairwise lncRNAs and mRNAs were calculated using the R program Hmisc (https://github.com/harrelfe/Hmisc). The significance of the differences between two correlation coefficients was assessed using the Fisher r-to-z transformation using an online calculator (http://vassarstats.net/rdiff.html). The one tail p value and z statistics was provided.
WGCNA correlation network analysis
To decrease the dataset size for network analysis, the lncRNA-mRNA pairs with |correlation coefficients| > = 0.7 were chosen for network analysis. Weighted correlation network analysis (WGCNA) including network construction, module detection, gene selection and calculations of topological properties was carried out to find clusters (modules) of highly correlated genes in the RNAseq datasets via R package WGCNA_1.49 [87]. One-step network construction and module detection methods were adopted in the analysis. A relatively large minimum module size of 30, and a medium sensitivity (deepSplit = 2) were applied for cluster splitting. The networks in each module with a TOM value > 0.5 were exported to an edge list file and plotted using the program VisANT [88]. We performed Overrepresentation test and GO annotation for mRNA gene sets using the online tool Pantherdb database (www.pantherdb.org), zebrafish genesets as reference and GO Slimmer (http://amigo1.geneontology.org/cgi-bin/amigo/slimmer). Fisher’s exact test was carried out with FDR multiple test correction and false discovery rate < 0.05. The expression heatmap was plotted using the software Genesis [89] with complete linkage mapping.
Differential expression analysis of stress-related and tissue-specific DE lncRNA genes
To characterize the stress-related lncRNAs in tilapia, the collected samples without replicates and stress challenges were firstly filtered. Finally, we kept 12 RNAseq samples in response to 24 h’s cold challenge that generated in this study and collected two published datasets under hypoxia and salinity challenges. To identify tissue-specific DE lncRNA genes, we randomly selected 12 tissues with three RNAseq datasets for each tissue. These datasets were then used to identify tissue-specific DE lncRNA genes by pair-wise comparisons of the different tissues.
The differential expression analysis for the DE lncRNAs was performed using the tool run_DE_analysis.pl that implemented in the trinityrnaseq-2.0.6 program [90, 91] with parameters setting as --method edgeR. The significant differentially expressed lncRNAs between test samples and control samples were defined when FDR (False Discovery Rate) was less than 0.05.
qRT-PCR analysis
qRT-PCR was performed as described in Xia et al. and Li et al. [31, 32]. Briefly, the amplifications were performed on the Roche Light Cycler 480 Real-time PCR System in a total volume of 10 μl (including 5 μl of 2 X SYBR Green MasterMix reagent, 1 μl of 1:10 diluted cDNA and 0.2 μl of each primer (10 μM)). The thermal cycling profile consisted of an initial denaturation at 95 °C for 5 min followed by 40 cycles of denaturation at 95 °C for 15 s, annealing at 60 °C for 15 s and extension at 72 °C for 20 s. An additional temperature-ramping step from 95 °C to 65 °C was used to produce the melting curve. All reactions were conducted in triplicate and using the gene EF-1α as internal control. The expression levels of genes were normalized by the levels of EF-1α in the same sample. Two-side t-test was used to compare expression levels.
LncRNA-microRNA interaction
The RNA interaction analysis was conducted with the microRNA target prediction tool miRanda (ver.3) [54] and the dre and fru miRNA datasets that downloaded from the miRBase (www.mirbase.org). Only matched miRNA-lncRNA pairs with minimum free energy (MFE) < − 12 kcal/mol and the miRanda total scores exceeding 120 were considered as significant pairs, as suggested in Hsu et al... [55].
References
Penny GD, Kay GF, Sheardown SA, Rastan S, Brockdorff N. Requirement for Xist in X chromosome inactivation. Nature. 1996;379:131.
Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10(3):155–9.
Ma L, Bajic VB, Zhang Z. On the classification of long non-coding RNAs. RNA Biol. 2013;10(6):925–33.
Bartolomei MS, Zemel S, Tilghman SM. Parental imprinting of the mouse H19 gene. Nature. 1991;351(6322):153–5.
Hezroni H, Koppstein D, Schwartz MG, Avrutin A, Bartel DP, Ulitsky I. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep. 2015;11(7):1110–22.
Yamashita A, Shichino Y, Yamamoto M. The long non-coding RNA world in yeasts. Biochim Biophys Acta. 2016;1859(1):147–54.
Chen B, Zhang Y, Zhang X, Jia SL, Chen S, Kang L. Genome-wide identification and developmental expression profiling of long noncoding RNAs during Drosophila metamorphosis. Sci Rep. 2016;6:23330.
Tarifeno-Saldivia E, Valenzuela-Miranda D, Gallardo-Escarate C. In the shadow: the emerging role of long non-coding RNAs in the immune response of Atlantic salmon. Dev Comp Immunol. 2017;73:193–205.
Kaushik K, Leonard VE, Kv S, Lalwani MK, Jalali S, Patowary A, Joshi A, Scaria V, Sivasubbu S. Dynamic expression of long non-coding RNAs (lncRNAs) in adult zebrafish. PLoS One. 2013;8(12):e83616.
Arriaga-Canon C, Fonseca-Guzman Y, Valdes-Quezada C, Arzate-Mejia R, Guerrero G, Recillas-Targa F. A long non-coding RNA promotes full activation of adult gene expression in the chicken alpha-globin domain. Epigenetics. 2014;9(1):173–81.
Gopalakrishnan K, Kumarasamy S, Mell B, Joe B. Genome-wide identification of long noncoding RNAs in rat models of cardiovascular and renal disease. Hypertension. 2015;65(1):200–10.
Zhao Z, Bai J, Wu AW, Wang Y, Zhang J, Wang ZS, Li YS, Xu J, Li X. Co-LncRNA: investigating the lncRNA combinatorial effects in GO annotations and KEGG pathways based on human RNA-Seq data. Database (Oxford). 2015;2015. https://doi.org/10.1093/database/bav1082.
Tani H, Torimura M. Identification of short-lived long non-coding RNAs as surrogate indicators for chemical stress response. Biochem Biophys Res Commun. 2013;439(4):547–51.
Tani H, Onuma Y, Ito Y, Torimura M. Long non-coding RNAs as surrogate indicators for chemical stress responses in human-induced pluripotent stem cells. PLoS One. 2014;9(8):e106282.
Mizutani R, Wakamatsu A, Tanaka N, Yoshida H, Tochigi N, Suzuki Y, Oonishi T, Tani H, Tano K, Ijiri K, et al. Identification and characterization of novel genotoxic stress-inducible nuclear long noncoding RNAs in mammalian cells. PLoS One. 2012;7(4):e34949.
Wang XD, Lin JB, Li FH, Zhang C, Li JY, Wang CH, Dahlgren RA, Zhang HQ, Wang HL. Screening and functional identification of lncRNAs under beta-diketone antibiotic exposure to zebrafish (Danio rerio) using high-throughput sequencing. Aquat Toxicol. 2017;182:214–25.
Wang F, Li LP, Xu HM, Liu Y, Yang C, Cowley AW, Wang NS, Liu PY, Liang MY. Characteristics of long non-coding RNAs in the brown Norway rat and alterations in the Dahl salt-sensitive rat. Sci Rep. 2014;4(1):7146.
Su S, Liu J, He K, Zhang MY, Feng CH, Peng FY, Li B, Xia XM. Overexpression of the long noncoding RNA TUG1 protects against cold-induced injury of mouse livers by inhibiting apoptosis and inflammation. FEBS J. 2016;283(7):1261–74.
Gracey AY, Fraser EJ, Li W, Fang Y, Taylor RR, Rogers J, Brass A, Cossins AR. Coping with cold: an integrative, multitissue analysis of the transcriptome of a poikilothermic vertebrate. Proc Natl Acad Sci U S A. 2004;101(48):16970–5.
Long Y, Song GL, Yan JJ, He XZ, Li Q, Cui ZB. Transcriptomic characterization of cold acclimation in larval zebrafish. BMC Genomics. 2013;14:612.
Long Y, Li L, Li Q, He X, Cui Z. Transcriptomic characterization of temperature stress responses in larval zebrafish. PLoS One. 2012;7(5):e37209.
Ju Z, Dunham RA, Liu Z. Differential gene expression in the brain of channel catfish (Ictalurus punctatus) in response to cold acclimation. Mol Gen Genomics. 2002;268(1):87–95.
Podrabsky JE, Somero GN. Changes in gene expression associated with acclimation to constant temperatures and fluctuating daily temperatures in an annual killifish Austrofundulus limnaeus. J Exp Biol. 2004;207(13):2237–54.
Kassahn KS, Crozier RH, Ward AC, Stone G, Caley MJ. From transcriptome to biological function: environmental stress in an ectothermic vertebrate, the coral reef fish Pomacentrus moluccensis. BMC Genomics. 2007;8:358.
Vornanen M, Hassinen M, Koskinen H, Krasnov A. Steady-state effects of temperature acclimation on the transcriptome of the rainbow trout heart. Am J Physiol Regul Integr Comp Physiol. 2005;289(4):R1177–84.
Gracey AY, Troll JV, Somero GN. Hypoxia-induced gene expression profiling in the euryoxic fish Gillichthys mirabilis. Proc Natl Acad Sci U S A. 2001;98(4):1993–8.
van der Meer DLM, van den Thillart GEEJM, Witte F, de Bakker MAG, Besser J, Richardson MK, Spaink HP, Leito JTD, Bagowski CP. Gene expression profiling of the long-term adaptive response to hypoxia in the gills of adult zebrafish. Am J Physiol-Reg I. 2005;289(5):R1512–9.
Marques IJ, Leito JT, Spaink HP, Testerink J, Jaspers RT, Witte F, van den Berg S, Bagowski CP. Transcriptome analysis of the response to chronic constant hypoxia in zebrafish hearts. J Comp Physiol B. 2008;178(1):77–92.
Ton C, Stamatiou D, Liew CC. Gene expression profile of zebrafish exposed to hypoxia during development. Physiol Genomics. 2003;13(2):97–106.
Everett MV, Antal CE, Crawford DL. The effect of short-term hypoxic exposure on metabolic gene expression. J Exp Zool A Ecol Genet Physiol. 2012;317(1):9–23.
Xia JH, Li HL, Li BJ, Gu XH, Lin HR. Acute hypoxia stress induced abundant differential expression genes and alternative splicing events in heart of tilapia. Gene. 2018;639:52–61.
Li HL, Lin HR, Xia JH. Differential gene expression profiles and alternative isoform regulations in gill of Nile Tilapia in response to acute hypoxia. Marine Biotechnology (NY). 2017;19(6):551–62.
Paneru B, Al-Tobasei R, Palti Y, Wiens GD, Salem M. Differential expression of long non-coding RNAs in three genetic lines of rainbow trout in response to infection with Flavobacterium psychrophilum. Sci Rep. 2016;6:36032.
Fao RF: FAO yearbook: fishery and aquaculture statistics, 2015. 2017.
Hu P, Liu M, Liu YM, Wang JF, Zhang D, Niu HB, Jiang SW, Wang J, Zhang DS, Han BS, et al. Transcriptome comparison reveals a genetic network regulating the lower temperature limit in fish. In: Scientific reports, vol. 6; 2016. p. 28952.
Zhao Y, Wang J, Thammaratsuntorn J, Wu JW, Wei JH, Wang Y, Xu JW, Zhao JL. Comparative transcriptome analysis of Nile tilapia (Oreochromis niloticus) in response to alkalinity stress. Genet Mol Res. 2015;14(4):17916–26.
Xu Z, Gan L, Li TY, Xu C, Chen K, Wang X, Qin JG, Chen LQ, Li EC. Transcriptome profiling and molecular pathway analysis of genes in association with salinity adaptation in Nile Tilapia Oreochromis niloticus. PLoS One. 2015;10(8):e0136506.
Li HL, Gu XH, Li BJ, Chen X, Lin HR, Xia JH. Characterization and functional analysis of hypoxia-inducible factor HIF1alpha and its inhibitor HIF1α in tilapia. PLoS One. 2017;12(3):e0173478.
Li HL, Gu XH, Li BJ, Chen CH, Lin HR, Xia JH. Genome-wide QTL analysis identified significant associations between hypoxia tolerance and mutations in the GPR132 and ABCG4 genes in Nile Tilapia. Mar Biotechnol. 2017;19(5):441–53.
Gu XH, Jiang DL, Huang Y, Li BJ, Chen CH, Lin HR, Xia JH. Identifying a major QTL associated with salinity tolerance in Nile Tilapia using QTL-Seq. In: Marine biotechnology; 2018.
Zhao Y, Zhu CD, Yan B, Zhao JL, Wang ZH. miRNA-directed regulation of VEGF in tilapia under hypoxia condition. Biochem Biophys Res Commun. 2014;454(1):183–8.
Zhao Y, Wu JW, Wang Y, Zhao JL. Role of miR-21 in alkalinity stress tolerance in tilapia. Biochem Biophys Res Commun. 2016;471(1):26–33.
Yan B, Zhao LH, Guo JT, Zhao JL. miR-429 regulation of osmotic stress transcription factor 1 (OSTF1) in tilapia during osmotic stress. Biochem Biophys Res Commun. 2012;426(3):294–8.
Yan B, Guo JT, Zhao LH, Zhao JL. MiR-30c: a novel regulator of salt tolerance in tilapia. Biochem Biophys Res Commun. 2012;425(2):315–20.
Wang B, Gan Z, Cai SH, Wang ZL, Yu DP, Lin ZW, Lu YS, Wu ZH, Jian JC. Comprehensive identification and profiling of Nile tilapia (Oreochromis niloticus) microRNAs response to Streptococcus agalactiae infection through high-throughput sequencing. Fish Shellfish Immunol. 2016;54:93–106.
Qiang J, Tao YF, He J, Xu P, Bao JW, Sun YL. miR-122 promotes hepatic antioxidant defense of genetically improved farmed tilapia (GIFT, Oreochromis niloticus) exposed to cadmium by directly targeting a metallothionein gene. Aquat Toxicol. 2017;182:39–48.
Qiang J, Tao YF, He J, Li HX, Xu P, Bao JW, Sun YL. Inhibition of miR-92d-3p enhances inflammation responses in genetically improved farmed tilapia (GIFT, Oreochromis niloticus) with Streptococcus iniae infection by modulating complement C3. Fish Shellfish Immunol. 2017;63:367–75.
Huang CW, Li YH, Hu SY, Chi JR, Lin GH, Lin CC, Gong HY, Chen JY, Chen RH, Chang SJ, et al. Differential expression patterns of growth-related microRNAs in the skeletal muscle of Nile tilapia (Oreochromis niloticus). J Anim Sci. 2012;90(12):4266–79.
Luo HL, Yang HZ, Lin Y, Zhang YD, Pan CY, Feng PF, Yu YL, Chen XH. LncRNA and mRNA profiling during activation of tilapia macrophages by HSP70 and Streptococcus agalactiae antigen. Oncotarget. 2017;8(58):98455–70.
Ling H, Spizzo R, Atlasi Y, Nicoloso M, Shimizu M, Redis RS, Nishida N, Gafà R, Song J, Calin GA. CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res. 2013;23(9):1446–61.
Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25(18):1915–27.
Wang SH, Ge W, Luo ZX, Guo Y, Jiao BL, Qu L, Zhang ZY, Wang X. Integrated analysis of coding genes and non-coding RNAs during hair follicle cycle of cashmere goat (Capra hircus). BMC Genomics. 2017;18:767.
Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22(9):1775–89.
Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003;5(1):R1.
Hsu PW, Huang HD, Hsu SD, Lin LZ, Tsou AP, Tseng CP, Stadler PF, Washietl S, Hofacker IL. miRNAMap: genomic maps of microRNA genes and their target genes in mammalian genomes. Nucleic Acids Res. 2006;34(Database issue):D135–9.
Xiao HM, Yuan ZT, Guo DH, Hou BF, Yin CL, Zhang WQ, Li F. Genome-wide identification of long noncoding RNA genes and their potential association with fecundity and virulence in rice brown planthopper, Nilaparvata lugens. BMC Genomics. 2015;16:749.
Wu YQ, Cheng TC, Liu C, Liu DL, Zhang Q, Long RW, Zhao P, Xia QY. Systematic identification and characterization of long non-coding RNAs in the silkworm, Bombyx mori. PLoS One. 2016;11(1):e0147147.
Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer Discovery. 2011;1(5):391–407.
Zhou ZY, Li AM, Adeola AC, Liu YH, Irwin DM, Xie HB, Zhang YP. Genome-wide identification of long intergenic noncoding RNA genes and their potential association with domestication in pigs. Genome Biol Evol. 2014;6(6):1387–92.
Jenkins AM, Waterhouse RM, Muskavitch MA. Long non-coding RNA discovery across the genus anopheles reveals conserved secondary structures within and beyond the Gambiae complex. BMC Genomics. 2015;16:337.
Young RS, Marques AC, Tibbit C, Haerty W, Bassett AR, Liu JL, Ponting CP. Identification and properties of 1,119 candidate lincRNA loci in the Drosophila melanogaster genome. Genome Biol Evol. 2012;4(4):427–42.
Sytnikova YA, Rahman R, Chirn GW, Clark JP, Lau NC. Transposable element dynamics and PIWI regulation impacts lncRNA and gene expression diversity in Drosophila ovarian cell cultures. Genome Res. 2014;24(12):1977–90.
Brown JB, Boley N, Eisman R, May GE, Stoiber MH, Duff MO, Booth BW, Wen J, Park S, Suzuki AM, et al. Diversity and dynamics of the Drosophila transcriptome. Nature. 2014;512(7515):393–9.
Ruan J, Dean AK, Zhang W. A general co-expression network-based approach to gene expression analysis: comparison and applications. BMC Syst Biol. 2010;4:8.
Liao Q, Liu CN, Yuan XY, Kang SL, Miao RY, Xiao H, Zhao GG, Luo HT, Bu DC, Zhao HT, et al. Large-scale prediction of long non-coding RNA functions in a coding-non-coding gene co-expression network. Nucleic Acids Res. 2011;39(9):3864–78.
Hao YB, Wu W, Shi FC, Dalmolin RJS, Yan M, Tian F, Chen XB, Chen GY, Cao W. Prediction of long noncoding RNA functions with co-expression network in esophageal squamous cell carcinoma. BMC Cancer. 2015;15:168.
Ghazalpour A, Doss S, Zhang B, Wang S, Plaisier C, Castellanos R, Brozell A, Schadt E, Drake T, Lusis A, et al. Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet. 2006;2(8):e130.
Raymond CS, Murphy MW, O'Sullivan MG, Bardwell VJ, Zarkower D. Dmrt1, a gene related to worm and fly sexual regulators, is required for mammalian testis differentiation. Genes Dev. 2000;14:2587–95.
Adelman CA, Petrini JH. ZIP4H (TEX11) deficiency in the mouse impairs meiotic double strand break repair and the regulation of crossing over. PLoS Genet. 2008;4:e1000042.
Mallon BS, Chenoweth JG, Johnson KR, Hamilton RS, Tesar PJ, Yavatkar AS, Tyson LJ, Park K, Chen KG, Fann YC, et al. StemCellDB: the human pluripotent stem cell database at the National Institutes of Health. Stem Cell Res. 2013;10(1):57–66.
Roth RB, Hevezi P, Lee J, Willhite D, Lechner SM, Foster AC, Zlotnik A. Gene expression analyses reveal molecular relationships among 20 regions of the human CNS. Neurogenetics. 2006;7(2):67–80.
Valenzuela-Miranda D, Gallardo-Escarate C. Novel insights into the response of Atlantic salmon (Salmo salar) to Piscirickettsia salmonis: interplay of coding genes and lncRNAs during bacterial infection. Fish Shellfish Immunol. 2016;59:427–38.
Wang XD, Zheng YS, Zhang YN, Li JY, Zhang HQ, Wang HL. Effects of beta-diketone antibiotic mixtures on behavior of zebrafish (Danio rerio). Chemosphere. 2016;144:2195–205.
Frigault JJ, Lang-Ouellette D, Morin P Jr. Up-regulation of long non-coding RNA TUG1 in hibernating thirteen-lined ground squirrels. Genomics Proteomics Bioinformatics. 2016;14(2):113–8.
Lu Y, Zhao X, Liu Q, Li C, Graves-Deal R, Cao Z, Singh B, Franklin JL, Wang J, Hu H, et al. lncRNA MIR100HG-derived miR-100 and miR-125b mediate cetuximab resistance via Wnt/beta-catenin signaling. Nat Med. 2017;23(11):1331–41.
Jiang L, Shao C, Wu QJ, Chen G, Zhou J, Yang B, Li H, Gou LT, Zhang Y, Wang Y, et al. NEAT1 scaffolds RNA-binding proteins and the microprocessor to globally enhance pri-miRNA processing. Nat Struct Mol Biol. 2017;24(10):816–24.
Augoff K, McCue B, Plow EF, Sossey-Alaoui K. miR-31 and its host gene lncRNA LOC554202 are regulated by promoter hypermethylation in triple-negative breast cancer. Mol Cancer. 2012;11:5.
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta stone of a hidden RNA language? Cell. 2011;146(3):353–8.
Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465(7301):1033–8.
Arvey A, Larsson E, Sander C, Leslie CS, Marks DS. Target mRNA abundance dilutes microRNA and siRNA activity. Mol Syst Biol. 2010;6:363.
Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–11.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and , Pachter L: Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc 2012, 7:562–578.
Mount DW. Using the Basic Local Alignment Search Tool (BLAST). CSH protocols. 2007;2007:pdb top17.
Li AM, Zhang JY, Zhou ZY. PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinformatics. 2014;15:311.
Sun L, Luo H, Bu D, Zhao G, Yu K, Zhang C, Liu Y, Chen R, Zhao Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013;41(17):e166.
Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhang SQ, Wei L, Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35:W345–9.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559.
Hu ZJ, Mellor J, Wu J, DeLisi C. VisANT: an online visualization and analysis tool for biological interaction data. BMC Bioinformatics. 2004;5:17.
Sturn A, Quackenbush J, Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics. 2002;18(1):207–8.
Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31(1):46–53.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52.
Acknowledgements
We thank one anonymous reviewer for comments on the manuscript and Dr. Wan Ziyi for helping in editing English language.
Funding
This work was supported by the National Natural Science Foundation of China (No.31572612); Science and Technology Planning Project of Guangdong Province, China (2017A030303008); Science and Technology Program of Guangzhou, China (No. 201803020043).
Availability of data and materials
All the read data under cold stress were available at the DDBJ database (BioProject Accession: PRJDB6721). All other datasets have been presented within the manuscript and/or additional supporting files.
Author information
Authors and Affiliations
Contributions
JHX and HRL contributed to project conception. BJL and DLJ collected the materials. BJL, DLJ, ZNM and YZ conducted the experiments. Data analysis was conducted by: BJL, DLJ, ZNM, YZ and JHX. The manuscript was prepared by JHX and BJL. HRL critically reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experiments in this study were approved by the Animal Care and Use Committee of the School of Life Science at Sun Yat-Sen University and were performed according to the regulations and guidelines established by this committee.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Summary information for RNAseq data and Transcriptome Shotgun Assembly (TSA) databases used in this study. (XLSX 18 kb)
Additional file 2:
Figure S1. The length distribution of the lncRNAs identified in this study. (TIF 915 kb)
Additional file 3:
Figure S2. The gene expression clustering of the RNAseq samples from 12 tissues used in the WGCNA analysis. (TIFF 1416 kb)
Additional file 4:
Table S2. Summary for the top hub genes with TOM values more than 0.5 in modules from 12 tissue RNAseq dataset. (XLSX 2060 kb)
Additional file 5:
Figure S3. An example showing VisANT visualization of the WGCNA weighted network. The network presents the relationships among the top hub genes in the ‘grey60” module for the RNAseq dataset. Only lncRNA-mRNA pair with TOM values > = 0.8 were presented in the network. Green circle: mRNAs; and blue circle: lncRNAs. (TIF 463 kb)
Additional file 6:
Table S3. Overrepresentation test of the lncRNA-associated mRNA geneset in Pantherdb database. Annotation was conducted using PANTHER version 13.1 Released 2018-02-03, Fisher’s exact with FDR multiple test correction and false discovery rate < 0.05, using Danio rerio (REF) as background geneset. (ODS 22 kb)
Additional file 7:
Table S4. Primer information used in the validation of DE transcript expression by qRT-PCR. (XLSX 11 kb)
Additional file 8:
Table S5. Summary for the DE lncRNA genes detected in tilapia in response to cold, salt and hypoxia stress. (XLSX 19 kb)
Additional file 9:
Table S6. Summary for tissues-specific DE lncRNA genes in tilapia. (XLSX 174 kb)
Additional file 10:
Figure S4. An heatmap example showing expression changes (Log2 transformed) of partial lncRNAs among tissues as indicated by the RNAseq data. The tissue type for each sample was shown above the heatmap. (TIF 691 kb)
Additional file 11:
Table S7. The interaction between miRNAs and lncRNA data identified in tilapia. (XLSX 575 kb)
Additional file 12:
72,276 lncRNA sequences identified in tilapia genome.rar. (FNA 56998 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, B.J., Jiang, D.L., Meng, Z.N. et al. Genome-wide identification and differentially expression analysis of lncRNAs in tilapia. BMC Genomics 19, 729 (2018). https://doi.org/10.1186/s12864-018-5115-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-5115-x