
Abstract
Background
Mutant peptides presented by MHC (major histocompatibility complex) Class II in cancer are important targets for cancer immunotherapy. Both animal studies and clinical trials in cancer patients showed that CD4 T cells specific to tumor-derived mutant peptides are essential for the efficacy of immune checkpoint blockade therapy by PD1 antibody.
Results
In this study, we analyzed the next generation sequencing data of 147 lung adenocarcinoma patients from The Cancer Genome Atlas and predicted neoantigens presented by MHC Class I and Class II molecules. We found 18,175 expressed clonal somatic mutations, with an average of 124 per patient. The presentation of mutant peptides by an HLA(human leukocyte antigen) Class II molecule, HLA DRB1, were predicted by NetMHCIIpan3.1. 8804 neo-peptides, including 375 strong binders and 8429 weak binders were found. For HLA DRB1*01:01, 54 strong binders and 896 weak binders were found. The most commonly mutated genes with predicted neo-antigens are KRAS, TTN, RYR2, MUC16, TP53, USH2A, ZFHX4, KEAP1, STK11, FAT3, NAV3 and EGFR.
Conclusions
Our results support the feasibility of discovering individualized HLA Class II presented mutant peptides as candidates for immunodiagnosis and immunotherapy of lung adenocarcinoma.
Similar content being viewed by others


Background
The efficacy of therapeutic effect of immune checkpoint blockade such as PD1 and CLTA4 antibodies is hypothesized to be dependent on mutant peptide epitopes which cause the T cell dependent cytotoxicity toward tumor cells. Epitopes for CD4 T cells are proposed to be a major mechanism. In mouse models, both artificial protein antigens and mutant peptide antigens derived from tumor cells were found to elicit tumorcidal T cell responses [1,2,3]. Clinical trials using long peptides or mRNA to deliver CD4 T cell epitopes to dendritic cells have shown success in inducing mutant peptide-specific CD4 T cells and their association with anti-tumor efficacy [4,5,6].
In this study, we analyzed next generation sequencing data from 147 lung adenocarcinoma patients deposited in the Cancer Genome Atlas, to identify both the driver and passenger mutations which may be presented by HLA Class II molecules. Due to the complexity of polymorphisms of both alpha and beta chains of HLA Class II molecules, we only studied the binding of mutant peptides to HLA DRB1 molecules that pair with an invariant alpha chain, HLA DRA.
Methods
Standardization and tracking of mutation data from TCGA
We collected mutations of lung adenocarcinoma from TCGA [7]. The data collection criteria was established as follows: 1, Tumor and matched normal adjacent tissue were included; 2, Samples that contain all somatic mutation, expression, SNP (single nucleotide polymorphism) array information were included; 3, Tumor samples from same patients were removed; 4, Samples with purity lower than 20% or ploidy larger than 6 were removed, purity and ploidy were reported by AbsCN-seq [8].
To remove common sequencing artifacts or residual germ line variation, each mutation was subjected to a ‘Panel of Normals’ filtering process using a panel of over 600 BAM files from normal samples. Mutations observed more than 1% in the panel of normals, dbSNP [9] or 1000G [10] were removed. Finally, all mutations with covered reads less than 10X were filtered out.
Purity and ploidy analysis
Purity and ploidy were estimated by AbsCN-seq, a software developed for WES (whole exon sequencing) data, based on SNV (single nucleotide variations) frequency and segment copy number.
Mutation clonality analysis
After estimating the tumor purity, we calculated the CCF (cancer cell fraction) for each mutation. The CCF is the percentage of tumor cells harboring a given mutation. Clonal mutations have a true CCF of 1, and subclonal mutations have a true CCF < 1. The observed allele counts correspond to a probability density of the CCF, which can be estimated with the following equation, where q(m) is the local copy number at the given mutation m, a is purity, and CCF ranges from 0 to 1. pdf is probability density function, alt is the alternate allele counts, ref. is the reference allele counts [11].
Neo-peptides prediction
We first confirmed that the mutated genes were expressed by RNA-seq data. Genes with 3 or more reads covered were defined as expressed according to Kandoth et al. [12]. 29-mer polypeptides centered on mutated residues were scanned to identify candidate peptides binding to MHC Class I or II molecules [13], i.e., peptide sequences surrounding mutated amino acids resulting from missense mutations, frame-shift or non-frame-shift indels. The affinity of 8–11 peptides binding to MHC Class I molecules were predicted using the NetMHCPan2.4 binding algorithm [14]. The affinity of 15 mer peptides binding to MHC Class II molecules were predicted using the NetMHCIIPan3.1 binding algorithm [15]. Threshold for strong binding peptides is defined as half-maximum inhibitory concentration (IC50) < 50 nM; Threshold for weak binding peptides is defined as IC50 < 500 nM [15,16,17].
MHC Class II molecules include HLA DP, DQ, and DR molecules. These molecules are composed of alpha and beta subunits. For DP and DQ molecules, both alpha and beta subunits are polymorphic. DR molecules are composed by a polymorphic beta subunit and an invariant alpha subunit. In this study, we focused on HLA DRB1, the most prevalent beta subunit of HLA DR [18]. The frequencies of other DRB molecules (DRB3, 4 and 5) are 5 to 10 fold lower than DRB1 (reference [18]). Clearly DRB1 molecules are significantly more frequent in presenting neo-antigens.
Results
To ensure high quality mutation calls for lung adenocarcinoma, stringent filters (Methods) were applied in sample and mutation collecting. A total of 40,229 somatic mutations in 147 lung adenocarcinomas were included for downstream analysis, including 26,296 missense, 8965 silent, 2061 nonsense, 911 splice site, 98 non-stop/read through, 1735 frame shift insertions/deletions (indels) and 163 inframe indels.
We assessed the CCF(cancer cell fraction) of each mutation as described in Carter et al. [19] to assess whether mutations are clonal (i.e., present in all cancer cells). Mutations are considered clonal if the CCF is close to 1. To determine the CCF, we calculated the sample purity (i.e., the percentage of tumor cells in sample), ploidy (i.e., a measure of the number of chromosomes in a cell) and absolute copy number by Abs-CNseq. We further identified clonal mutations based on beta distribution. In total, we identified 21,710 clonal mutations (Fig. 1), including the known proliferation-related genes (e.g., TP53, KRAS, EGFR).

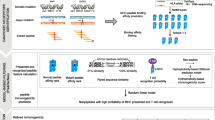
Flow chart of clonal mutation analysis and HLA-binding neo-antigen prediction for lung adenocarcinoma patients
High-affinity candidate T cell epitopes were identified in silico by scanning of the mutant peptides resulting from missense mutations, frame-shift or non-frame-shift indels. T cell epitopes presented by MHC Class I molecules were predicted by NetMHCPan2.4 binding algorithm (Additional file 1: Table S1, Additional file 2: Table S2 and Additional file 3: Table S3). T cell epitopes presented by MHC Class II molecules were predicted by NetMHCIIPan3.1 binding algorithm. We focused on HLA DRB1, the most prevalent beta subunit of HLA DR which pairs with invariant alpha subunit HLA DRA [18]. In total, 8804 neo-peptides, including 375 strong binders and 8429 weak binders were found (Fig. 2). For DRB1*01:01, 950 neo-peptides, including 54 strong binders and 896 weak binders were found. The most commonly mutated genes with predicted neo-antigens are KRAS, TTN, RYR2, MUC16, TP53, USH2A, ZFHX4, KEAP1, STK11, FAT3, NAV3 and EGFR (Table 1). The exact mutated sequences are listed in Additional file 4: Table S4. The frequency of neo-peptides varies widely in individual patients of lung adenocarcinomas, from 0 to 523 (Fig. 2). Table 2 shows the distribution of neo-antigens in different HLA DRB1 alleles. DRB1*01:02, DRB1*12:01, DRB1*11:04, DRB1*01:01 were found to be the most frequent DRB1 alleles which present neo-antigens. High frequency of neo-peptides were found in hotspots of KRAS (Table 3, G12C or G12 V). INDEL mutations were found in most patients (Fig. 3). However, no linear correlation was found between SNV and INDEL mutations.

Predicted HLA-DRB1-binding neo-antigen mutant peptides in 147 lung adenocarcinoma patients. Somatic mutations were predicted by NetMHCIIPan3.1. All patients were lined up according to numbers of HLA-DRB1-binding neo-antigen mutations, including both strong-binders (SB, blue color) and weak-binders (WB, red color). Gray color indicates other mutations which do not bind to MHC Class II molecules. Smokers and non-smokers were analyzed separately

Predicted HLA-DRB1-binding INDEL mutant peptides in 147 lung adenocarcinoma patients
Discussion
Several groups have proposed to predict HLA Class II presented neo-antigens through next generation sequencing for cancer immunotherapy [1,2,3,4,5,6]. In both mouse models and human patients, the function of predicted neo-antigens have been verified,by measuring CD4 T cell responses or tumor rejection.
In this study, we have predicted the HLA Class II-presented neo-antigen peptides in lung adenocarcinoma. An average of 59 HLA DRB1-presented neo-antigen mutations were predicted per lung cancer patient. This prediction is based on the assumption that all HLA DRB1 alleles may be the MHC class II molecule to present mutated peptides in a patient. Since a specific cancer patient only express one HLA DRB1 allele, the actual mutant peptide epitope presented by a cancer patient is much lower. Unfortunately, the HLA DRB1 allele data are not available in public TCGA database for the lung cancer patients we have studied. Assuming HLA DRB1*01:01 is the HLA DRB1 allele, 54 strong binders and 896 weak binders were found in 147 patients. In average, 5 mutant peptides were found per patient with HLA DRB1*01:01 allele.
van Buuren et al. reported that the sensitivity of neo-epitope prediction from analysis of exonic SNVs in cancer exome sequencing data requires little improvement [20]. Our analysis on mutant peptides presented by HLA Class I molecules in lung cancer patients is consistent with this conclusion (Additional file 1: Table S1 and Additional file 5: Table S5, top mutated genes with predicted epitopes binding to HLA Class I molecules).
A weakness of our analysis is that the expression of predicted neo-epitopes could not be determined. As we described, genes with 3 or more reads covered in RNA-seq data were defined as expressed according to Kandoth et al. [12]. Although the normal copy of a gene may be expressed, its variants may not be expressed, especially truncating variants that may undergo nonsense-mediated transcript decay. Mass spectrometry-based new technologies are emerging to verify predicted neo-epitopes [21,22,23], through analysis of eluted peptides from HLA molecules purified from cancer tissues.
K-Ras, TP53, and EGFR mutants are well known vaccine candidates which are currently in clinical trials [24,25,26,27]. Our data suggest that such mutations in proliferation-related genes are also candidate for CD4 epitopes. In addition, neo-antigens of passenger mutations are also attractive targets for individualized precision therapy. There is urgent need for technologies which may help to determine whether the predicted neo-antigen mutations are presented by HLA Class II molecules. Technical platforms include ELISPOT assay by synthetic candidate peptide epitopes, T cell stimulation assay by using antigen presenting cell lines expressing specific HLA DRB1 molecules, and tetramer staining-based sorting of neoantigen-specific T cells.
Conclusions
This study used clonal mutation analysis to predict HLA DRB1 molecule presented neo-antigen mutant peptides which are expressed at RNA level. Genes discovered here provide clues for identifying CD4 T cell epitopes for immune monitoring and therapy.
Abbreviations
- CCF:
-
Cancer cell fraction
- EGFR:
-
Epidermal growth factor receptor
- ELISPOT:
-
Enzyme-Linked ImmunoSpot
- HLA:
-
Human leukocyte antigen
- MHC:
-
Major histocompatibility complex
- SNP:
-
Single nucleotide polymorphism
- SNV:
-
Single nucleotide variations
- TCGA:
-
The Cancer Genome Atlas
- WES:
-
Whole exon sequencing
References
Sun Z, Chen F, Meng F, Wei J, Liu B. MHC class II restricted neoantigen: a promising target in tumor immunotherapy. Cancer Lett. 2017;392:17–25.
Flament H, Alonso Ramirez R, Prémel V, Joncker NT, Jacquet A, Scholl S, Lantz O. Modeling the specific CD4+ T cell response against a tumor neoantigen. J Immunol. 2015;194(7):3501–12. https://doi.org/10.4049/jimmunol.1402405. Epub 2015 Mar 2. Erratum in: J Immunol. 2017 Aug 15;199(4):1526
Urban JL, Schreiber H. Tumor antigens. Annu Rev Immunol. 1992;10:617–44.
Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, Menn O, Osswald M, Oezen I, Ott M, Keil M, Balß J, Rauschenbach K, Grabowska AK, Vogler I, Diekmann J, Trautwein N, Eichmüller SB, Okun J, Stevanović S, Riemer AB, Sahin U, Friese MA, Beckhove P, von Deimling A, Wick W, Platten M. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. 2014;512(7514):324–7.
Nielsen JS, Chang AR, Wick DA, Sedgwick CG, Zong Z, Mungall AJ, Martin SD, Kinloch NN, Ott-Langer S, Brumme ZL, Treon SP, Connors JM, Gascoyne RD, Webb JR, Berry BR, Morin RD, Macpherson N, Nelson BH. Mapping the human T cell repertoire to recurrent driver mutations in MYD88 and EZH2 in lymphoma. Oncoimmunology. 2017;6(7):e1321184.
Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, Chen C, Olive O, Carter TA, Li S, Lieb DJ, Eisenhaure T, Gjini E, Stevens J, Lane WJ, Javeri I, Nellaiappan K, Salazar AM, Daley H, Seaman M, Buchbinder EI, Yoon CH, Harden M, Lennon N, Gabriel S, Rodig SJ, Barouch DH, Aster JC, Getz G, Wucherpfennig K, Neuberg D, Ritz J, Lander ES, Fritsch EF, Hacohen N, Wu CJ. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–21.
Lander ES. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–50.
Bao L, Pu M, Messer K. AbsCN-seq: a statistical method to estimate tumor purity, ploidy and absolute copy numbers from next generation sequencing data. Bioinformatics. 2014;30(8):1056–63.
Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–11.
Abecasis GR, David A, Adam A, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–73.
Brastianos PK, Amaro TW, Manley PE, et al. Exome sequencing identifies BRAF mutations in papillary craniopharyngiomas. Nat Genet. 2014;46(2):161–5.
Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333.
Sahin U, Derhovanessian E, Miller M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222.
Morten N, Claus L, Thomas B, et al. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS One. 2007;2(8):e796.
Nielsen M, Justesen S, Lund O, et al. NetMHCIIpan-2.0 - Improved pan-specific HLA-DR predictions using a novel concurrent alignment and weight optimization training procedure. Immunome Res. 2010;6(1):9.
Hoof I, Peters B, Sidney J, et al. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61(1):1.
Turajlic S, Litchfield K, Xu H, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. 2017;18(8):1009.
Gragert L, Madbouly A, Freeman J, Maiers M. Six-locus high resolution HLA haplotype frequencies derived from mixed-resolution DNA typing for the entire US donor registry. Hum Immunol. 2013;74:1313–20.
Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30(5):413–21.
van Buuren MM, Calis JJ, Schumacher TN. High sensitivity of cancer exome-based CD8 T cell neo-antigen identification. Oncoimmunology. 2014;3:e28836. eCollection 2014
Bassani-Sternberg M, Bräunlein E, Klar R, Engleitner T, Sinitcyn P, Audehm S, Straub M, Weber J, Slotta-Huspenina J, Specht K, Martignoni ME, Werner A, Hein R, H Busch D, Peschel C, Rad R, Cox J, Mann M, Krackhardt AM. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat Commun. 2016;7:13404.
Polyakova A, Kuznetsova K, Moshkovskii S. Proteogenomics meets cancer immunology: mass spectrometric discovery and analysis of neoantigens. Expert Rev Proteomics. 2015;12(5):533–41.
Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER, Linette GP. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348(6236):803–8.
Ebben JD, Lubet RA, Gad E, Disis ML, You M. Epidermal growth factor receptor derived peptide vaccination to prevent lung adenocarcinoma formation: an in vivo study in a murine model of EGFR mutant lung cancer. Mol Carcinog 2015. https://doi.org/10.1002/mc.22405. [Epub ahead of print].
Li G, Wong AJ. EGF receptor variant III as a target antigen for tumor immunotherapy. Expert Rev Vaccines. 2008;7(7):977–85.
Hartley ML, Bade NA, Prins PA, Ampie L, Marshall JL. Pancreatic cancer, treatment options, and GI-4000. Hum Vaccin Immunother. 2015;11(4):931–7.
Chaft JE, Litvak A, Arcila ME, Patel P, D'Angelo SP, Krug LM, Rusch V, Mattson A, Coeshott C, Park B, Apelian DM, Kris MG, Azzoli CG. Phase II study of the GI-4000 KRAS vaccine after curative therapy in patients with stage I-III lung adenocarcinoma harboring a KRAS G12C, G12D, or G12V mutation. Clin Lung Cancer. 2014;15(6):405–10.
Funding
National Natural Science Foundation of China grant 81570007; National Key Research and Development Plan grant 2017YFA050590.
Availability of data and materials
Raw sequencing data of genome, exome and transcriptome can be downloaded at TCGA data portal ( https://portal.gdc.cancer.gov) by disease category LUAD. Raw data were retrieved from public domain as follows:
(1) Somatic Mutations https://tcga-data.nci.nih.gov/docs/publications/luad_2014/AN_TCGA_LUAD_PAIR_capture_freeze_FINAL_230.aggregated.capture.tcga.uuid.curated.somatic.maf
(5) Clinical information of samples https://tcga-data.nci.nih.gov/docs/publications/luad_2014/TCGA_LUAD_Clinical_Info.xlsx
Author information
Authors and Affiliations
Contributions
WC, DZ, WW, WLT, JW, CZ, and YL designed the study, performed data analysis, and wrote the paper. All authors approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Top mutated genes with predicted HLA Class I binding neo-peptides in 147 lung adenocarcinoma patients in this study. T cell epitopes presented by MHC Class I molecules were predicted by NetMHCPan2.4 binding algorithm. (XLSX 192 kb)
Additional file 2:
Table S2. Number of predicted neo-antigen peptides presented by MHC Class I molecules in 147 lung adenocarcinoma patients. T cell epitopes presented by MHC Class I molecules were predicted by NetMHCPan2.4 binding algorithm. MHC-I molecules which are significantly more frequent in presenting neo-antigens were labelled as bold according to P values. Significant levels were calculated using one sided Mann-Whitney U test. (XLSX 11 kb)
Additional file 3:
Table S3. Amino acid sequences of predicted MHC class I binding neo-peptides of KRAS, EGFR, TP53, and MUC16 in 147 lung adenocarcinoma patients in this study. T cell epitopes presented by MHC Class I molecules were predicted by NetMHCPan2.4 binding algorithm. (XLSX 13 kb)
Additional file 4:
Table S4. Amino acid sequences of predicted MHC Class II molecule HLA DRB1 binding neo-peptides in 147 lung adenocarcinoma patients in this study. (XLSX 503 kb)
Additional file 5:
Table S5. Amino acid sequences of predicted MHC class I binding neo-peptides in 147 lung adenocarcinoma patients in this study. (XLSX 534 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cai, W., Zhou, D., Wu, W. et al. MHC class II restricted neoantigen peptides predicted by clonal mutation analysis in lung adenocarcinoma patients: implications on prognostic immunological biomarker and vaccine design. BMC Genomics 19, 582 (2018). https://doi.org/10.1186/s12864-018-4958-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-4958-5