
Abstract
Background
Bovine milk is an important nutrient source for humans. Forage plays a vital role in dairy husbandry via affecting milk quality and quantity. However, the differences in mammary metabolism of dairy cows fed different forages remain elucidated. In this study, we utilized transcriptomic RNA-seq and iTRAQ proteomic techniques to investigate and integrate the differences of molecular pathways and biological processes in the mammary tissues collected from 12 lactating cows fed corn stover (CS, low-quality, n = 6) and alfalfa hay (AH, high-quality, n = 6).
Results
A total of 1631 differentially expressed genes (DEGs; 1046 up-regulated and 585 down-regulated) and 346 differentially expressed proteins (DEPs; 138 increased and 208 decreased) were detected in the mammary glands between the CS- and AH-fed animals. Expression patterns of 33 DEPs (18 increased and 15 decreased) were consistent with the expression of their mRNAs. Compared with the mammary gland of AH-fed cows, the marked expression changes found in the mammary gland of CS group were for genes involved in reduced mammary growth/development (COL4A2, MAPK3, IKBKB, LGALS3), less oxidative phosphorylation (ATPsynGL, ATP6VOA1, ATP5H, ATP6VOD1, NDUFC1), enhanced lipid uptake/metabolism (SLC27A6, FABP4, SOD2, ACADM, ACAT1, IDH1, SCP2, ECHDC1), more active fatty acid beta-oxidation (HMGCS1), less amino acid/protein transport (SLC38A2, SLC7A8, RAB5a, VPS18), reduced protein translation (RPS6, RPS12, RPS16, RPS19, RPS20, RPS27), more proteasome- (PSMC2, PSMC6, PSMD14, PSMA2, PSMA3) and ubiquitin-mediated protein degradation (UBE2B, UBE2H, KLHL9, HSPH1, DNAJA1 and CACYBP), and more protein disassembly-related enzymes (SEC63, DNAJC3, DNAJB1, DNAJB11 and DNAJC12).
Conclusion
Our results indicate that the lower milk production in the CS-fed dairy cows compared with the AH-fed cows was associated with a network of mammary gene expression changes, importantly, the prime factors include decreased energy metabolism, attenuated protein synthesis, enhanced protein degradation, and the lower mammary cell growth. The present study provides insights into the effects of the varying quality of forages on mammary metabolisms, which can help the improvement of strategies in feeding dairy cows with CS-based diet.
Similar content being viewed by others


Background
Forage accounts for 50% or more of the diet of dairy cows [1]. Therefore, the quality of forage has a large effect on bovine milk production in the dairy industry. In China, alfalfa hay (AH) is widely used as a high-quality forage, but approximately 230 million kilogram of AH has to be imported from other countries annually because of a shortage [2], substantially increasing milk production cost. Furthermore, the gap between supply and demand for high-quality forage is increasing by approximately 10% annually [3]. However, China produces approximately 100 million tons of corn stover (CS; a main crop byproduct produced in North China) each year, and most of this crop byproduct is disposed or burned [4]. The use of CS in dairy feed has been limited due to its low nutritional value. From an environmental prospective and to reduce the dependence on imported AH, it is strategically important for China to explore how to efficiently utilize these low quality forages in dairy production. Therefore, it is our goal to investigate the regulatory mechanism through which various forages exert their effects on dairy production, which will help us to develop new methods to improve the efficiency of utilizing low-quality forages (such as CS) in dairy feeding.
Compared with AH, CS has lower protein content (crude protein, rumen degradable protein, and rumen ungradable protein) and non-fiber carbohydrates [5]. In recent studies from our group, cows fed AH-based diets had better production performance in milk yield, milk protein content, and milk efficiency (milk yield/dry matter intake) compared to cows fed CS-based diets [6]. To explore the mechanisms underlying the low production performance of cows fed a CS diet, we performed the following studies. First, we found that the dairy cows fed an AH-based diet had enhanced metabolisms of several amino acids, including phenylalanine, serine, threonine, tyrosine, arginine and proline, in the mammary gland compared to cows consuming CS-based diets [7]. Second, we demonstrated that low feed and nitrogen efficiency may play a vital role in contributing to low milk protein production in the mammary glands of cows consuming low-quality forage of CS and rice straw [8]. However, the regulatory mechanisms by which the different-quality forages affect bovine mammary metabolisms and the subsequent milk production remained largely unknown.
Sustained developments in the nucleotide sequencing technology, especially RNA-sequencing, has resulted in an explosive growth in the number and quality of transcriptome sequenced for various tissues in both prokaryotes and eukaryotes [9, 10] and their potential changes under different conditions. Also, a certain of dynamicity by post-transcriptional and translational regulations of gene expression can be estimated by measuring its proteome (expressed protein set from genome) [9]. To date, iTRAQ-based quantitative proteomics has extensively improved protein identification coverage, thus providing more comprehensive linking of proteins to their metabolic functions. Recently, integrating transcriptomic and quantitative proteomic analyses have been widely used to promote a better understanding of the molecular mechanisms driving biological process in cells and tissues [11,12,13,14,15]. Here our objective was to elucidate a more complete understanding of molecular mechanisms underlying mammary gland adaptation to the alternative nutrient supplies provided by two rations with different forage sources (AH and CS). We performed transcriptomic and proteomic analyses of the mammary gland tissue samples collected from 12 lactating cows fed either AH) or CS).
Methods
Experiment design and sample collection
All animal care and procedures in this study were approved by the Animal Care Committee, Zhejiang University (Hangzhou, P. R. China) and were in accordance with the Zhejiang University’s guidelines for animal research. Twelve multiparous Chinese Holstein dairy cows were randomly assigned into two blocks, among which six cows with the milk yield of 30.3 ± 5.1 kg/d (mean ± SD) and the body weight of 604 ± 37.4 kg were fed CS as forage, and the other six cows with the milk yield of 29.2 ± 4.4 kg/d and the body weight of 608 ± 41.4 kg were fed AH as forage. The two blocks of cows were fed 55% concentrate (dry matter, DM), 15% corn silage, and either 30% CS (CS group; n = 6) or 23% AH and 7% Chinese wild rye hay (AH group; n = 6). The dietary compositions and nutritional values of both diets were reported previously [6]. The experiment was conducted over a 14-wk period, with the first 2 wk. for dietary adaptation, in Hangjiang Dairy Farm (Hangzhou, China). The cows were slaughtered at the end of experiment, and mammary tissue samples were collected from each cow, placed in sterile tubes, immediately snap-frozen in liquid N2, and subsequently stored at − 80 °Cfor extraction of RNA and protein.
RNA preparation, cDNA synthesis and RNA sequencing
RNA was extracted from mammary tissue with Trizol reagent (Invitrogen, Carlsbad, CA, USA) and purified with Qiagen RNeasy kit (Qiagen, Valencia, CA, USA). RNA quality was examined with the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA), and RNA quantity was measured with a NanoDrop (NanoDrop Technologies, Inc. Wilmington, DE, USA). The RNA integrity number (RIN) [16] was more than 7.0. Approximately 5 μg of high quality RNA was used for cDNA library construction according to the Illumina RNA ligation-based method [17]. 2.5 μg RNA from the mammary gland of two cows in each group were combined (three biological replicates per group; Fig. 1), and RNA sequencing was performed using a HISeq 2000 sequencing system (Illumina, San Diego, CA, USA) from LC Sciences (Houston, TX, USA). Reads were processed with Bowtie version 0.12.7 (http://bowtie-bio.sourceforge.net), aligned in Tophat version 1.3.2 (http://ccb.jhu.edu/software/tophat/index.shtml), and then mapped to bovine reference genome UMD3.1 (ftp://ftp.ensembl.org/pub/release-79/fasta/bos_taurus/dna/) [18]. Gene abundance was estimated as reads per kilobase of exon model per million mapped reads (RPKM) with Cufflinks version 1.2.1 as previously described [17, 18]. Genes with a cut-off of 1.5-fold expression changes and p-value less than 0.05 between CS and AH groups were defined as differentially expressed.

An overview of the transcriptomic and proteomic experiment. Schematic diagram of workflow of the RNA-seq transcriptomic and iTRAQ-based proteomic experiments. Six cows were fed either corn stover (CS) or alfalfa hay (AH) as forage for 14 weeks. RNA from two cows and protein samples from three cows within each group were pooled for transcriptomic (3 replicates/group) and proteomic (2 replicates/group) analyses. For transcriptomic assay, the pooled RNA was sequenced on the Illumina platform, and subsequently the reads were aligned and mapped to the Bos taurus genome. For the proteomic assay, the extracted proteins were digested with trypsin, and the peptides are labeled with different iTRAQ reagents, which contain reporter groups of different masses (114, 115, 116, 117), balance groups of different masses (191, 190, 189, 188), and a reactive group (R). The labeled peptides are then mixed equivalently and fractionated by strong cation exchange (SCX) chromatography. Fractions were separated by liquid chromatography (LC) and analyzed by two-step mass spectrometry (MS)
The qRT-PCR analysis
Total RNA extracted from the mammary gland was reverse transcribed for cDNA synthesis using a PrimeScriptRT Reagent Kit with gDNA Eraser (Takara, Tokyo, Ostu, Japan) following the manufacturer’s instructions. The qRT-PCR was performed in triplicate using the Applied Biosystems 7500 real-time PCR system (Applied Biosystems, Foster City, CA, USA). Each 20 μL reaction included 50 ng of reverse transcription product, 40 nM of each forward and reverse primer [Additional file 1: Table S1, designed by Primer 5 software (Premier Biosoft International, Palo Alto, CA, USA)], and SYBR Premix Taq (Takara). The PCR program was one cycle of 95 °C for 30 s plus 40 cycles of amplification at 95 °C for 5 s and 58 °C for 30 s, followed by an additional 15 s at 95 °C, 1 min at 60 °C, and 15 s at 95 °C to generate melt curves. The relative gene expression values were calculated by the 2−ΔΔCt method [19]. The gene expression levels were normalized against the internal control genes β-actin and GAPDH.
Protein preparation and digestion
Protein preparation and digestion were performed as in the previous studies [20, 21]. Briefly, 500 mg mammary tissue was ground to a fine powder in liquid N2, lysed with the lysis buffer A (7 M Urea, 2 M Thiourea, 4% CHAPS (3-[(3-Cholamidopropy) dimethylammonio] propane-sulfonate), 40 mM Tris-HCl, pH 8.5), and reduced with 10 mM DTT at 56 °C for 1 h, followed by alkylation with 55 mM IAM (Iodoacetamide) in a darkroom for 1 h. The reduced and alkylated protein mixtures were precipitated by adding 4 × volume of chilled acetone and incubating at − 20 °C overnight. After centrifugation at 4 °C and 30,000×g, the pellet was dissolved in 0.5 M TEAB (Triethylamine borane; Applied Biosystems, Milan, Italy) and sonicated on ice. After centrifugation at 30,000×g at 4 °C again, an aliquot of the supernatant was assigned for determination of protein concentration by the Bradford method [22]. The proteins in the supernatant were kept at − 80 °C for further analysis.
The iTRAQ labeling and strong cationic exchange (SCX) fractionation
Total protein (150 μg obtained by mixing 50 μg protein from the mammary glands of three cows in each group, two biological replicates per group; Fig. 1) was digested with Trypsin Gold (Promega, Madison, WI, USA) at 37 °C for 16 h with the ratio of protein: trypsin = 30: 1. After digestion, peptides were dried by vacuum centrifugation. Peptides were then reconstituted in 0.5 M TEAB and processed following the manufacturer’s protocol for 4-plex iTRAQ reagent (Applied Biosystems) [21]. The protein samples from the CS and AH groups were labeled with iTRAQ reagents 114, 115, 116 and 117. Strong cationic exchange chromatography was performed with an LC-20AB HPLC pump system (Shimadzu, Kyoto, Japan). The procedures for SCX fractionation including the elution were essentially the same as in the study of Meng et al. [20]. Finally, the eluted peptides were pooled into 20 fractions, desalted with a Strata X C18 column (Phenomenex, Torrance, CA, USA), and vacuum-dried.
Liquid chromatography–tandem mass spectrometry (LC/MS) analysis
The sample fractions described above were further separated and identified on an LC-20 AD nano-HPLC system (Shimadzu) loaded with Q-Extractive mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA). Buffer C consisted of 2% acetonitrile (ACN) and 0.1% formic acid (FA) in Milli-Q water; buffer D consisted of 98% ACN and 0.1% FA. After resuspension with buffer C, 10 μL sample supernatant was loaded by the auto-sampler onto a C18 trap column (2 cm × 100 μm, 5 μm) and then separated on the reverse-phase analytical C18 column (100 mm × 75 μm, 3 μm). The samples were loaded at 8 μL/min for 4 min, then a 44 min gradient was run at 300 nL/min starting from 2 to 35% buffer D, followed by 2 min linear gradient to 80% buffer D, and then maintenance at 80% buffer D for 4 min, and finally return to 5% buffer D in 1 min.
Peptide analysis was performed with a Q-Exactive mass spectrometer in a positive ion mode with a selected mass range of 350–2000 mass/charge (m/z). The electrospray voltage applied was 1.6 kV. For MS scans, the m/z scan range was 350 to 2000 Da. For MS/MS scans, the m/z scan range was 100–1800. MS/MS data was acquired using the top 15 most abundant precursor ions with the ion count more than 20,000 in the MS scan. These were selected with an isolation window of 2 m/z and were fragmented via high energy collisional dissociation under normalized collision energies of 30 eV. For the MS scan, the resolving power was set to 70,000 at m/z 200, the maximal ion injection time was 10 ms and, dynamic exclusion of the selected precursor ions was 15 s. Automatic gain control (AGC) was used to optimize the spectra generated from the orbitrap, and the AGC target value was 3,000,000 for full MS and 100,000 for MS2, respectively. For the MS/MS scans, the resolving power was set to 17,500 at m/z 200; maximum ion injection times for MS/MS scans were at 60 ms; and the underfill ratio was defined as 0.1%.
Protein identification and quantification
The raw files were first merged and transformed to an MGF file with Proteome Discoverer ver. 1.2 (Thermo Fisher Scientific, San Jose, CA, USA) and were then probed on the Mascot search engine (ver. 2.3.02; Matrix Science, London, UK) of the Uniprot database of bovine (Bos tauru) with 31,661 entries. For protein identification, the parameters were set the same as in the study by Yang et al. [23] with some minor changes: carbamidomethyl (C), iTRAQ 4-plex (N-term), and iTRAQ 4-plex (K) were defined as fixed modifications; Gln→pyro-Glu (N-term Q), oxidation (M), deamidated (NQ) were the possible variable modifications. The decoy database pattern was considered as the reverse of the target database. The method of peptide identification by false discovery rate (FDR) was performed as Sheng et al. described [24].
Relative quantification of the identified proteins was performed with the Proteome Discoverer software described above and calculated by the weighted and normalized ratios of uniquely identified peptides that belong to the specific individual protein. The integration window tolerance of the peak was set to 20 ppm. Statistics analysis was conducted using Fisher’s test. Proteins with a cutoff of 1.2-fold change between CS and AH samples and p < 0.05 were determined as significantly differentially expressed proteins.
Bioinformatics analysis
Functional annotations were performed using Blast2GO program against the non-redundant protein database (NR; NCBI). Metabolic pathway analysis was conducted using R software (R version 3.2.3) according to the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database (http://www.genome.jp/kegg/). The pathway enrichment statistics were performed by Fisher’s exact test with a p-value ≤ 0.05 considered as significant.
Western blot analysis
Approximately 40 μg protein per sample was separated on 12% SDS (sodium dodecyl sulfate) polyacrylamide gels. Proteins were transferred onto 0.45 μm PVDF membranes (IPVH00010; Millipore, Boston, Massachusetts, USA) and blocked with blocking buffer (Beyotime, Jiangsu, China). The membranes were incubated with primary antibodies to SCP2 (non-specific lipid-transfer protein 2, ab140126; Abcam, Cambridge, MA, USA), IDH2 (isocitrate dehydrogenase 2; ab131263, Abcam), SLC7A8 [(solute carrier family 7 (amino acid transporter, L system), member 8, ab75610, Abcam)], COL4A2 (collagen, type IV, alpha 2, sc-70,243, Santa Cruz biotechnology; Cambridge, MA, USA), and β-actin (Beyotime). After washing with TBST [tris-buffered saline containing 0.02% (v/v) Tween-20] three times, the membranes were incubated with goat anti-rabbit IgG or goat anti-mouse IgG secondary antibodies conjugated with horseradish peroxidase (Beyotime), incubated with ECL (electrochemiluminescence) Western Blotting Substrate Kits (Beyotime), and finally visualized with a Kodak Image Station 2000MM (Kodak Molecular Imaging Systems, New Haven, USA). The relative intensities of bands were calculated with ImagePro Plus 6.0 software (Media Cybernetics, Washington, MD, USA) using β-actin as the reference protein.
Statistical analysis
The data on relative mRNA expression by qRT-PCR and protein expression by Western blot were analyzed through one-way ANOVA in SAS (SAS 9.0). The statistical significance was declared at p ≤ 0.05.
Results
Overview of transcriptomic and quantitative proteomic analyses
Figure 1 shows the workflow of our integrative RNA-seq transcriptomic and iTRAQ-based proteomic experiments. In the transcriptomic analysis, 78,138,798 and 90,522,588 raw/clean reads were detected in the CS and AH groups, respectively (Additional file 2: Table S2), and in proteomic analysis, 62,367 unique spectra were strictly matched to 24,606 unique peptides and further mapped to 3744 unique proteins (Additional file 2: Table S3). With a cutoff of 1.5-fold change and a p-value < 0.05, a total of 1631 differentially expressed genes (DEGs) were identified between CS and AH groups in transcriptomic analysis, among which 1046 DEGs were up-regulated and 585 were down-regulated (Fig. 2 and Additional file 3: Table S4). Using a threshold of 1.2-fold change and p < 0.05, 346 differentially expressed proteins (DEPs; 138 up-regulated and 208 down-regulated) were detected between CS and AH groups in proteomic analysis (Fig. 2 and Additional file 3: Table S5). By comparing the RNA-seq data with the proteomic data (Fig. 2 and Additional file 3: Table S6), 40 genes displayed differential expression at both mRNA and protein levels, of which 18 and 15 genes were consistently up- or down-regulated, respectively, whereas the remaining 7 genes had inconsistent expression in mRNA and protein levels, which may result from post-translational modifications.

The venn diagram of the differentially expressed genes (DEGs) and proteins (DEPs) in the mammary gland of cows fed either corn stover (CS) or alfalfa hay (AH). The cut-off of differential expression of mRNA is set at 1.5-fold change and p < 0.05, whereas the cutoff of differential expression of protein is set at 1.2-fold and p < 0.05
The gene ontology (GO) analysis of DEGs and DEPs
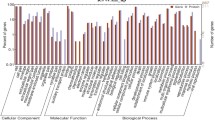
The GO analysis of all the DEGs and DEPs in cows fed CS versus AH is shown in Fig. 3. In the cellular component category, most of the DEGs and DEPs were mainly assigned to cell (25.8% genes and 24% proteins), cell part (25.8% genes and 24% proteins), organelle (18.9% genes and 19.2% proteins) and organelle part (9.8% genes and 10.2% proteins). Notably, a small number of the DEGs and DEPs were located in the extracellular region (3.9% genes and 5.4% proteins), membrane-enclosed lumen (3.9% genes and 3.0% proteins) and macromolecular complexes (6.5% genes and 6.9% proteins). In the biological process category, a large number of DEGs and DEPs were involved in cellular process (16.9% genes and 17.2% proteins), metabolic process (13.0% genes and 12.6% proteins) and biological regulation (10.9% genes and 9.0% proteins). Noticeably, some DEGs and DEPs were assigned to response to stimulus (4.8% genes and 7.3% proteins), immune system process (2.3% genes and 2.4% proteins), cellular component biogenesis (2.3% genes and 2.3% proteins) and cell growth (0.8% genes and 0.9% proteins). For the molecular function category, the GO terms including binding (43.1% genes and 41.7% proteins), enzyme regulator activity (29.1% genes and 5.1% proteins) and catalytic activity (16.6% genes and 30.6% proteins) were the predominant functions of the DEGs and DEPs, and a relatively low proportion of DEGs and DEPs were associated with various biological activities such as molecular transducer (3.3% genes and 3.2% proteins), transcription regulator (2.9% genes and 1.4% proteins), transporter (2.2% genes and 7.8% proteins), structural molecule (1.6% genes and 6.0% proteins) and electron carrier (0.4% genes and 1.1% proteins). In general, the DEGs and DEPs displayed similar GO annotation patterns.

Gene ontology (GO) categories assigned to the differentially expressed genes (DEGs, inner cycle) and proteins (DEPs, outer cycle) in the mammary gland of cows fed either corn stover (CS) or alfalfa hay (AH). The differentially expressed genes were classified into cellular component, biological process, and molecular function by WEGO (Web Gene Ontology Annotation Plot) according to the GO terms
We also performed further functional analysis of the up- and down-regulated genes and proteins using the UniProt knowledgebase and GO database. The increased genes (Fig. 4) in the CS group were significantly and abundantly enriched for genes involved in the positive regulation of apoptosis, unfolded protein binding, negative regulation of protein ubiquitination and response to stress. In addition, we found that some genes with increased abundance were enriched in the negative regulations of translation, DNA binding, NF-κB transcription factor activity, phosphorylation, cell cycle and in proteasome-mediated/ubiquitin-dependent protein catabolic processes. Notably, nine genes with up-regulated expression were enriched in the GO term GDP binding. In contrast, the decreased genes (Fig. 4) were principally enriched in cell adhesion, positive regulation of cell proliferation, binding activities of actin/receptor/carbohydrate, structural molecule activity and protein binding (295 decreased genes enriched). Additionally, some down-regulated genes were related to the positive regulation of several transcriptional factors (Stat3 phosphorylation and NF-κB signaling) and cell-growth associated processes (cell proliferation, multicellular organism growth, and cell growth). Importantly, a small number of decreased genes were associated with the cytoskeleton/extracellular matrix and cytokine-mediated signaling pathway. Notably, four decreased genes were significantly enriched in the GO term “cellular response to amino acid stimulus”. Furthermore, the GO term “ATPase activity” was significantly enriched in six down-regulated genes.

Functional characterization of the increased or decreased transcripts in the mammary gland of cows fed alfalfa hay (AH) vs. corn stover (CS) by gene ontology analysis. DEG indicates differentially expressed genes. The x-axis shows the functional categories of the increased or decreased genes, the left y-axis shows the value of –Log (p-value) and the right y-axis shows the number of increased/decreased genes
The up-regulated proteins (Fig. 5) were primarily enriched in negative regulation of multicellular organismal and developmental processes. In addition, we observed a small proportion of increased proteins enriched in the GO terms related to lipid translocation including lipid transport and long-chain fatty acid binding, and in GO terms involved in cation homeostasis and ion channel inhibitor activity. Specifically, eight up-regulated proteins were enriched in enzyme inhibitor activity, which may suggest a general reduction of the enzyme-mediated metabolism. The GO annotation analysis of the down-regulated proteins is shown in Fig. 5. Notably, the most abundant GO term was extracellular matrix organization enriched in ten down-regulated proteins. In addition, the body defense-related GO terms, such as response to external stimulus, cellular response to amino acid stimulus, and defense response, were significantly enriched with some decreased proteins. Some proteins of down-regulation were involved in tissue development, organ morphogenesis, biological adhesion and response to transforming growth factor beta. Importantly, 16 down-regulated proteins were involved in substrate-specific transporter activity. Furthermore, six decreased proteins were enriched in glycosyl-compound biosynthetic process and three decreased proteins were related to ATPase activity.

Functional characterization of the increased or decreased proteins in the mammary gland of cows fed alfalfa hay (AH) vs. corn stover (CS) by gene ontology analysis. DEP indicates differentially expressed proteins. The x-axis shows the functional categories of increased or decreased proteins, the left y-axis shows the value of –Log (p-value) and the right y-axis shows the number of increased/decreased proteins
KEGG pathway analysis of DEGs and DEPs
Table 1 shows the KEGG pathway enrichment analysis of DEGs. In the analysis, a total of 7 decreased DEGs were significantly enriched in the pathway of protein digestion and absorption (p = 0.0269). In contrast, 10 increased DEGs were significantly enriched in protein processing in endoplasmic reticulum (p = 0.045). Additionally, we found that 4, 11, and 7 increased genes were significantly enriched in protein degradation-related processes ubiquitin mediated proteolysis, spliceosome and proteasome, respectively. In particular, the significant pathways (p ≤ 0.05) enriched by some decreased DEGs were related to protein synthesis (such as ribosome biogenesis in eukaryotes, aminoacyl-tRNA biosynthesis and lysosome). Also, a small number of DEGs were significantly enriched in the pathways of mTOR signaling pathway, p53 signaling pathway, and two cell-growth-associated signaling pathways (the TGF-beta signaling and Wnt signaling).
The KEGG pathway enrichment analysis of the DEPs is shown in Table 2, and a total of 29 KEGG pathways were significantly enriched. Importantly, 7 decreased DEPs were significantly enriched (p = 0.0101) in the “protein digestion and absorption” pathway, while 7 increased DEPs were significantly enriched (p = 0.037) in the “protein processing in endoplasmic and reticulum” pathway. Several DEPs were involved in pathways associated with energy metabolism― glycolysis/gluconeogenesis (p = 0.0302), citrate cycle (p = 0.0399), pentose phosphate pathway (p = 0.0352), and PI3K-Akt signaling (p = 0.0375). Additionally, the KEGG pathways enriched by a small number of DEPs were related to the protein synthesis/processing (including ribosome, p = 0.0069; aminoacyl-tRNA biosynthesis, p = 0.0236; mTOR signaling pathway, p = 0.035; lysosome, p = 0.0350; protein processing in endoplasmic reticulum, p = 0.0370; and spliceosome, p = 0.0232) and the metabolisms of several amino acids. Noticeably, we also found that a certain of proteins were significantly enriched in two cell-growth-signaling pathways (Wnt and TGF-beta signaling pathways; p = 0.0375 and 0.0272, respectively).
Functional analysis of the common DEGs and DEPs
The 40 common expressed genes at mRNA and protein levels were further analyzed by the GO and KEGG pathway (Additional file 4: Table S7). Importantly, 3 increased genes at mRNA and protein levels (DnaJ homolog subfamily B members—DNAJB11, DNAJB1, and DNAJC12) were related to protein unfolding and involved in the pathway of “protein processing in endoplasmic reticulum”. Also, two common genes of up-regulation (dihydrolipoamide branched chain transacylase, DBT and hydroxymethylglutaryl-CoA synthase, HMGCS1) were involved in the process of “valine, leucine and isoleucine degradation”. Noticeably, the decreased genes IDH2 and ATPase 2 (ATP2B4) at mRNA and protein levels were involved in TCA cycle and ATP synthesis, respectively; in contrast, 4 common genes of up-regulated expression [HMGCS1, SCP2, DBT and isopentenyl-diphosphate delta-isomerase 1 (IDI1))] were all associated with fatty-acyl-CoA metabolic process. In particular, the commonly decreased gene dimethylglycine dehydrogenase (DMGDH) was associated with “glycine, serine and threonine metabolism”. In addition, several commonly expressed genes of down-regulation [such as erythrocyte membrane protein (EPB41L3), odorant-binding protein (MGC151921), DMGDH, VPS18 protein (VPS18), and the uncharacterized protein ORAI1] were involved in the processes of “protein localization” and “transport”. Intriguingly, two up-regulated genes DHFR and KRT15 at mRNA and protein levels were related to one carbon metabolism and staphylococcus aureus infection, respectively. Additionally, the 3 collagens (COL1A1, COL1A2 and COL4A2) and mitogen-activated protein kinase 3 (MAPK3) down-regulated at both mRNA and protein levels were involved in the protein synthesis-related pathways of “protein digestion and absorption” and “ECM-receptor interaction”.
Verification of DEGs by qRT-PCR and DEPs by western blot analysis
Nineteen genes involved in energy metabolism, transcription/translation, protein processing/transport, protein degradation, amino acid metabolism/ transport, fatty acid oxidation, and mammary gland growth and development were selected for qRT-PCR analysis (Additional file 5: Table S8). Among these genes (Fig. 6), expression levels of 15 genes were significantly altered between CS- and AH- groups, and the abundance of 11 of the 15 gene abundance were consistent with expression patterns measured by RNA-seq. Four proteins, including IDH2, SLC7A8, SCP2, and COL4A2 were selected for Western blot analysis (Fig. 7 and Additional file 5: Table S8). Western blot analysis showed that the protein levels of IDH2, SLC7A8, and COL4A2 in the mammary glands of cows fed the CS-based diet were lower than those cows fed the AH diet, whereas the level of SCP2 was higher. All the immunoblot results were consistent with the findings from proteomic analysis.

Real time PCR analysis of mRNA expression changes of genes involved in mammary metabolism of cows fed corn stover (CS) and alfalfa hay (AH). Relative mRNA expression levels were normalized by the levels of β-actin. Error bars represent the standard deviation. ** and * indicate that the difference in gene expression between CS and AH groups reached p < 0.01, and 0.01 < p < 0.05, respectively

Western blot analysis of expression of IDH2, SLC7A8, SCP2, and COL4A2 proteins in the mammary gland of cows fed corn stover (CS) and alfalfa hay (AH). β-Actin was used as a sample loading control. ** and * indicate p < 0.01 and 0.01 < p < 0.05, respectively. IDH2: Isocitrate dehydrogenase 2; SLC7A8: also referred as LAT2, L type amino acid transporter 2; SCP2: sterol carrier protein 2; COL4A2: collagen type IV alpha 2
Relationship between the DEGs/DEPs and mammary metabolism
A comprehensive view of the molecular mechanisms underlying milk production was summarized based on the proteomic and transcriptomic data collected (Fig. 8 and Additional file 6: Table S9). The regulatory subsections proposed include: energy metabolism, amino acid (AA) /fatty acid metabolism, protein degradation, protein synthesis, protein processing, AA/protein transport, and cell growth and development. Each of these regulatory functions is involved in the regulation of mammary metabolism, and the collective analysis shows how the mammary gland adapts to the low nutrient availability of the CS ration compared with the higher nutrient availability on the AH ration. Despite that the gene- and protein-level responses are not always consistent, the relatively higher degree of agreement between analyses of DEGs and DEPs at the functional level suggests that transcriptomic analysis of the mammary gland might be sufficient to characterize tissue functional responses to altered states but might not always be reflective of shifts in specific proteins.

An overview of possible biological changes that might contribute to low milk production in cows fed corn stover-based diet vs. cows fed alfalfa hay-based diet. The color coding for the individual genes is as follows: black letters and pink background represents the up-regulated genes, yellow letters and pink background represents increased proteins, black letters and green background represents the down-regulated genes, and blue letters and green background represents decreased proteins. The half blue/half black letters with green background represents genes that were both down-regulated at mRNA and protein levels. The half black/half yellow letters with pink background represent the genes that were up-regulated at both protein and gene levels. The full name of each protein is listed in Additional file 6: Table S9
Discussion
Roughage is one of the most important components in ruminant diets, the quality of which has a large impact on milk performance in dairy cows [25]. Our previous study showed that compared to cows fed AH as the forage source, cows fed CS had lower yields of milk (kg/d; 23.5 vs. 19.4), milk fat (kg/d; 0.98 vs. 0.82), milk protein (kg/d; 0.77 vs. 0.62) and lactose (kg/d; 1.15 vs. 0.94) (P < 0.01) [6]. In addition, as a major crop byproduct, a large quantity of corn stover is burned by humans, and this inefficient utilization of crop residues subsequently produces heavy environmental pollution. Therefore, it is urgent for us to determine strategies to improve the utilization of low-quality forage (CS) in the dairy industry and reduce damage to the environment caused by burning CS. However, little is known about the effects of roughage consumption on the molecular mechanisms in milk production in the mammary gland. In this study, we detected 1046 up-regulated and 585 down-regulated genes by RNA-seq transcriptomics as well as 138 increased and 208 decreased proteins by iTRAQ proteomics in the mammary gland of dairy cows fed CS- versus AH-based diets. There were only 33 genes with consistent expression patterns measured by both transcriptomic and proteomic analyses, suggesting that the post-transcriptional regulation may play an important role in gene expression. The relatively limited overlap of proteins and mRNAs was also seen in other studies in human [14], mouse [11], fish [26] and bacteria [27].
The GO term annotation can help to characterize physiological and functional changes associated with the changes in mRNA and protein expression in cells and tissues [28]. In up-regulated genes in CS-fed cows versus AH-fed cows, the GO term “ubiquitin-dependent/proteasome-mediated protein catabolic process” was significantly enriched, which may indicate enhanced mammary protein catabolism in cows fed CS. Enrichment in the GO terms of negative regulation of translation, NF-κB transcription factor activity, and DNA binding activity suggested inhibited protein synthesis in the mammary gland, consistent with the reduced milk protein yield in CS-fed cows [6]. In addition, several proteins that were up-regulated in CS-fed cows were enriched in the GO term “enzyme inhibitor activity”, which is also consistent with an overall reduced mammary metabolism in these cows. Furthermore, some increased proteins were involved in positive regulation of apoptosis and negative regulations of multicellular organismal processes and developmental processes, in line with the reduced cell growth in the CS group as shown in our previous microRNAome analysis [8]. Interestingly, the fact that the GO terms of lipid transport and long-fatty acid binding were enriched in the up-regulated proteins in CS-fed cows supported the idea that the mammary gland of these cows may take up more fatty acids from the blood for mammary metabolism as these cows had a lower acetate supply to the mammary gland than AH-fed cows [6].
Consistently, the genes that were expressed lower in CS-fed cows were enriched for the GO terms of cell adhesion, positive regulation of cell proliferation, and multicellular organism growth, which also indicated the attenuated cell growth in the mammary gland of CS-fed animals. In addition, down-regulation of genes associated with positive regulation of Stat3 phosphorylation and NF-κB signaling pathway in CS-fed cows was consistent with the reduced protein synthesis in these animals. Meanwhile, the lower expression levels of the proteins involved in biological adhesion, extracellular matrix organization, tissue development, and organ morphogenesis were consistent with possible reduced mammary cell growth and mammary tissue development in CS-fed cows. Furthermore, the proteins that were lower in abundance were enriched in the GO term “response to transforming growth factor beta”, indicating that TGF-β may be a major signaling molecule in the regulation of mammary cell growth in cows fed CS. Collectively, GO analysis indicated that the reduced cell growth and metabolism, attenuated protein synthesis, and enhanced protein degradation may play major roles leading to the low milk production in CS-fed dairy cows.
The KEGG pathway analysis has also been widely used for systematic understanding of the gene functions in cells or organisms from large-scale molecular data sets [29, 30]. In this study, the up-regulated genes were enriched in ubiquitin mediated proteolysis, and the genes with lower abundance were enriched in ribosome (the main machinery for protein synthesis), suggesting that these pathways may play an integrative role in lowering milk protein production in the mammary gland of CS-fed cows [6]. Additionally, some down-regulated proteins from CS-fed cows were enriched in the mTOR signaling pathway and metabolisms of several amino acids, including glycine, serine, threonine, arginine, proline, phenylalanine, histidine, and tyrosine, which is also consistent with the lower milk protein in the CS group [6, 8]. In particular, three down-regulated proteins in CS-fed cows were associated with valine, leucine and isoleucine degradation, indicating a reduction of branched-amino acids production in milk [31]. Moreover, the pathways enriched among differentially expressed proteins included the Wnt and TGF-β signaling pathways. These pathways are known to play roles in regulating mammary growth and differentiation [32,33,34].
Milk production is a highly energy-dependent process and requires sufficient ATP [35]. In most eukaryotes, oxidative phosphorylation in the mitochondria involving a series of ATPases is the prime metabolic pathway to generate energy [36]. In the study, we observed that the expression of several ATPase components (including ATPsynGL, ATP6V0A1, ATP5H and ATP6V0D1) and one mitochondrial enzyme NADH dehydrogenase (ubiquinone) 1 subunit C1 (NDUFC1) involved in oxidative phosphorylation were lower in CS-fed cows. The lower levels of these enzymes may potentially contribute to a lower ATP production in the mammary tissue of these animals. For high-yield dairy cows, particularly during negative energy balance, ketone bodies (included acetoacetate, β-hydroxy-butyrate, and acetone) are produced in the liver but utilized in other tissues of the body (including the mammary gland) as an energy source [37]. Acetyl-CoA acetyltransferase (ACAT1) plays a major role in ketone body synthesis, transferring one acetyl group to another acetyl-CoA (the crucial substrate for ketone body generation) [38]. Furthermore, the rate-limiting enzyme in ketone body synthesis is HMGCS1, which promotes β-hydroxy-butyrate production [39]. The increased expression of both ACAT1 and HMGCS1 in CS-fed cows may reflect an increased level of mammary ketone body production in cows consuming CS, which results in an increased level of ketone body utilization as energy for the mammary gland of these cows, in line with the high abundance of β-hydroxybutyric acid and acetoacetyl-CoA in blood of CS-fed cows versus AH-fed cows [7].
Amino acids are fundamental proteogenic substrates for milk synthesis in dairy cows, most of which derive from dietary proteins. After digestion and absorption in the small intestine, amino acids are taken up from the blood by the mammary gland via amino acid transporters. The protein SLC7A8 is a system L amino acid transporter that functions by a Na+-independent and electroneutral transport mechanism for neutral amino acids [40]. In this study, mammary expression of SLC7A8 genes was 0.38- fold lower in CS-fed cows compared to the AH-fed cows, indicating a reduced system L amino acid transporter activity for neutral amino acids (including isoleucine, leucine, methionine, phenylalanine, threonine, alanine and serine) in the mammary gland of CS-fed cows [41]. This finding was in consistent with our previous observations of amino acid uptake in these cows [6, 31].
Ribosomes consist of a small 40S and a large 60S subunits, and ribosomal proteins are required for different stages of ribosome biogenesis and/or for distinct steps of the translation process [42]. All six ribosomal protein subunits (RPS2, RPS12, RPS16, RPS19, RPS20 and RPS27) expressed at lower levels in CS-fed cows were essential components of the small ribosomal subunit (40S). The lower abundance of these ribosomal proteins in the CS group suggests depressed efficiency of protein translation, contributing to lower milk protein in these cows [31].
For protein processing, proteins and peptides must be transported into the endoplasmic reticulum (ER) or Golgi, where they acquire modifications that allow them to be biologically active [43]. Specific three-dimensional conformations acquired through folding of newly translated polypeptides and/or refolding of unfolded proteins are essential for protein function formation and maintenance [44]. The protein DNAJB1 is a heat shock protein (HSP) [an ER molecular chaperone that can protect other proteins against occurrence of incorrect folding, but may also stimulate ER-associated degradation (ERAD)] [45]. As an ER molecular chaperone, the protein DNAJB11 is essential to prevent the processes such as protein aggregation, allowing protein folding and assembly to proceed correctly [46]. In this study, expression of DNAJB1 and DNAJB11 was higher at the mRNA and protein levels in the CS-fed cows, suggesting that these cows may have higher degree of protein non-aggregation and protein degradation in the mammary gland. The protein heat shock 105 kDa/110 kDa protein 1 (HSPH1), another HSP, can be induced by several kinds of environmental stress [47] and is associated with mammary tumor tissues [48]. In the current study, the highly levels of HSPH1 in the CS-fed cows may suggest that the CS diet induces ER-stress in the mammary gland. The increased amount of protein degradation in the mammary gland is in accordance with the reduced milk protein and lactation performance in the CS group [6].
The proteasome is a large, 26S-multicatalytic protease that degrades poly-ubiquitinated proteins to small peptides [49]. The proteasome is composed of two sub-complexes: a core catalytic 20S particle and a regulatory 19S particle [50]. Expression of two 19S regulatory particles [proteasome 26S subunit, ATPase 6 (PSMC6) and proteasome 26S subunit, ATPase 2 (PSMC2)], the two 20S core particles [proteasome subunit alpha 3 (PSMA3) and proteasome subunit alpha 2 (PSMA2)] and proteasome 26S subunit, non-ATPase 14 (PSMD14) was higher in the mammary gland of cows fed the CS-based diet, supporting enhanced protein degradation in these cows. In addition, many intracellular proteins become covalently modified with ubiquitin (UB) or ubiquitin-like proteins (UBLs) [51]. Then, ubiquitin-conjugating enzymes (E1s) transfers the activated modifier to a family of E2 ubiquitin-conjugating enzymes, leading to their degradation [52]. In this study, expression of ubiquitin-conjugating enzyme 2B (UBE2B), ubiquitin-conjugating enzyme E2H (UBE2H) and target recognizing subunit kelch-like family member 9 (KLHL9) was higher in CS-fed cows, further indicating active protein degradation in the mammary glands of these animals. Taken together, the higher abundance of the proteasome-related and ubiquitin-dependent proteins is consistent with a higher degree of protein degradation in the mammary cells of CS-fed cows. Both the possibly increased protein degradation and reduced protein synthesis may result in lower milk protein yield in the CS group.
Peroxisomes are essential organelles that play a key role in lipid homeostasis. The protein SCP2 acts in peroxisome cholesterol transport through the cytoplasm, and loss of SCP2 can result in defects in fatty acid β-oxidation [53]. Higher SCP2 measured at mRNA and protein levels in cows fed CS-based diet suggested an enhanced level of fatty acid β-oxidation in the mammary gland. The protein isocitrate dehydrogenase [NADP] 1 (IDH1) is an enzyme that catalyzes oxidative decarboxylation of isocitrate, producing alpha-ketoglutarate (α-ketoglutarate) and CO2. Cytosolic IDH1 (EC 1.1.1.41) and mitochondrial IDH2 (EC 1.1.1.42) catalyze the same reaction outside the context of the citric acid cycle and use NADP+ as a cofactor instead of NAD+ [54]. Cytosolic IDH1 plays a complementary role in reductive glutamine metabolism, possibly through its oxidative function in an IDH2/IDH1 shuttle that transfers high energy electrons in the form of NADPH from mitochondria to cytosol, especially under hypoxia [55]. In this study, the higher IDH1 and the lower levels of IDH2 in CS-fed cows may indicate that the complementary IDH1-dependent carboxylation pathway is enhanced in CS-fed cows, enabling higher citrate production for energy supply. Acyl-CoA dehydrogenases (ACADs) are a class of enzymes that function to catalyze the initial step in each cycle of fatty acid β-oxidation in the mitochondria of cells [56]. Higher expression of acyl-CoA dehydrogenase (ACADM), combined with the higher levels of long fatty acid transporter (SLC27A6), in CS-fed cows may indicate a higher transport activity of long fatty acid from the plasma into the mammary cells, thus supporting a higher level of fatty acid oxidation for energy supply in these cows. Taken together, expression changes of multiple genes involved in fatty acid β-oxidation and fatty acid transport indicate a more active fatty acid metabolism to supply energy in the CS-fed cows compared to AH-fed cows. This observation is consistent with our previous finding that the CS-fed cows had a lower supply of acetate which is the major energy source in ruminants [6].
Finally, six collagen proteins and five genes encoding collagen proteins were lower in the mammary gland of CS-fed cows. Collagens are the main structural proteins in extracellular matrix (ECM), and are important regulators of the differentiated phenotype of mammary epithelial cells in culture [31, 32]. Lower levels of these proteins may indicate a different mammary gland morphology in these animals. Specifically, a decrease in the levels of the protein COL4A2 was found to be associated with a loss of basement membrane integrity, accompanied by a dramatic alteration of alveolar morphology with decreased size and shrunken lumen containing little β-casein [32]. Mammary structural changes may be another factor contributing to the reduction of milk yield in the CS-fed cows compared with cows fed AH [31].
Conclusions
By integrating transcriptomic and proteomic data, this study suggested four major possible mechanisms contributing to the lower milk production in dairy cows fed CS-diet compared to AH-diet: (i) reduced mammary growth/development through lower expression of COL4A2, MAPK3, IKBKB, and LGALS3, (ii) less oxidative phosphorylation through lower expression of ATPsynGL, ATP6VOA1, ATP5H, ATP6VOD1, and NDUFC1 with enhanced lipid uptake and fatty acid beta-oxidation to supply energy through higher expression of SLC27A6, FABP4, SOD2, ACADM, ACAT1, IDH1, SCP2, ECHDC1, and HMGCS1, (iii) less AA/protein transport and metabolism through lower expression of SLC38A2, SLC7A8, RAB5a, and VPS18, and less protein translation through lower expression of RPS6, RPS12, RPS16, RPS19, RPS20, and RPS27, and (iv) more proteasome- and ubiquitin-mediated protein degradation through higher expression of the protease components PSMC2, PSMC6, PSMD14, PSMA2, PSMA3, and ubiquitin-conjugating enzymes UBE2B, UBE2H, KLHL9, HSPH1, DNAJA1, and CACYBP, and protein disassembly-associated enzymes SEC63, DNAJC3, DNAJB1, DNAJB11, and DNAJC12. These mechanisms involved in milk production of dairy cows fed CS-forage-based diet can direct future work to further understand how the mammary gland adapts to low nutrient availability and, ultimately, to change feeding strategies so that cows can utilize low-quality forage more efficiently.
Abbreviations
- AARS2 :
-
Probable alanyl-tRNA synthetase
- ACADM :
-
Acyl-CoA dehydrogenase, C-4 to C-12 straight chain
- ACAT1 :
-
Acetyl-CoA acetyltransferase
- ACN :
-
Acetonitrile
- ACSS1 :
-
Acetyl-coenzyme A synthetase 1
- AGC :
-
Automatic gain control
- AH :
-
Alfalfa hay based diet
- ATP5H :
-
ATP synthase subunit d
- ATP6V0A1 :
-
Isoform 1 of V-type proton ATPase 116 kDa subunit a isoform 1
- ATP6V0D1 :
-
V-type proton ATPase subunit d1
- ATPsynGL :
-
ATP synthase, H+ transporting, mitochondrial F0 complex, subunit G-like
- CACYBP :
-
Calcyclin binding protein
- CHAPS :
-
3-[(3-Cholamidopropy) dimethylammonio] propane-sulfonate
- COL4A2 :
-
Collagen, type IV, alpha 2
- CS :
-
Corn stover based diet
- DBT :
-
Dihydrolipoamide branched chain transacylase E2
- DMGDH :
-
Dimethylglycine dehydrogenase
- DNAJA1 :
-
DnaJ (Hsp40) homolog, subfamily A, member 1
- DNAJB1 :
-
DnaJ (Hsp40) homolog, subfamily B, member 1
- DNAJB11 :
-
DnaJ homolog subfamily B member 11
- DNAJC12 :
-
DnaJ homolog subfamily C member 12
- DNAJC3 :
-
DnaJ homolog subfamily C member 3
- ECHDC1 :
-
Enoyl CoA hydratase domain containing 1
- FA :
-
Formic acid
- FABP4 :
-
Fatty acid binding protein 4
- FDR :
-
False discovery rate
- GAPDH :
-
Glyceraldehyde-3-phosphate dehydrogenase
- HK1 :
-
Hexokinase 1
- HMGCS1 :
-
Hydroxymethylglutaryl-CoA synthase
- HSPH :
-
Heat shock 105 kDa/110 kDa protein 1
- HSPH1 :
-
Heat shock 105 kDa/110 kDa protein 1
- IDH1 :
-
Isocitrate dehydrogenase [NADP]
- IDH2 :
-
Isocitrate dehydrogenase [NADP]
- IKBKB :
-
Inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase beta
- KLHL9 :
-
Kelch-like family member 9
- LGALS3 :
-
Galectin-3-binding protein
- MAOA :
-
Amine oxidase [flavin-containing] A
- MAP K3 :
-
Mitogen-activated protein kinase 3
- NDUFC1 :
-
NADH dehydrogenase [ubiquinone] 1 subunit C1
- PDHX :
-
Pyruvate dehydrogenase complex, component X
- PFKP :
-
Phosphofructokinase
- POLR1A :
-
Polymerase (RNA) I polypeptide A, 194 kDa
- POLR1B :
-
Polymerase (RNA) I polypeptide B, 128 kDa
- POLR2B :
-
DNA-directed RNA polymerase
- POMP :
-
Proteasome maturation protein
- PSMA2 :
-
Proteasome subunit alpha 2
- PSMA3 :
-
Proteasome subunit alpha 3
- PSMC2 :
-
Proteasome 26S subunit, ATPase 2
- PSMC6 :
-
Proteasome 26S subunit, ATPase 6
- PSMD14 :
-
Proteasome 26S subunit, non-ATPase 14
- PYGB :
-
Phosphorylase, glycogen
- RAB5a :
-
Ras-related protein Rab-5A
- RPS12 :
-
40S ribosomal protein S12
- RPS16 :
-
40S ribosomal protein S16
- RPS19 :
-
40S ribosomal protein S19
- RPS2 :
-
40S ribosomal protein S2
- RPS20 :
-
40S ribosomal protein S20
- RPS27 :
-
40S ribosomal protein S27
- SARDH :
-
Sarcosine dehydrogenase
- SCP2 :
-
Sterol carrier protein 2
- SCX :
-
Strong cationic exchange
- SEC63 :
-
SEC63 homolog, protein translocation regulator
- SLC27A6 :
-
Solute carrier family 27 (fatty acid transporter), member 6
- SLC38A2 :
-
Solute carrier family 38, member 2
- SLC7A8 :
-
Solute carrier family 7 (amino acid transporter light chain, L system), member 8
- SOD2 :
-
Superoxide dismutase [Mn]
- SUCLG1 :
-
Succinyl-CoA ligase [GDP-forming] subunit alpha
- TARS2 :
-
Threonyl-tRNA synthetase 2
- TEAB :
-
Triethylamine borane
- UBE2B :
-
Ubiquitin-conjugating enzyme E2B
- UBE2H :
-
Ubiquitin-conjugating enzyme E2H
- VPS18 :
-
VPS18 protein
References
Kendall C, Leonardi C, Hoffman P, Combs D. Intake and milk production of cows fed diets that differed in dietary neutral detergent fiber and neutral detergent fiber digestibility. J Dairy Sci. 2009;92(1):313–23.
Wang JQ. Five key indicators of leading the direction of China dairy industry. Feed and Husbandry. 2011;4:003.
Li XL, Wan LQ. Research progress on Medicago sativa silage technology [J]. Acta Pratac Sci. 2005;2:001.
Pang Y, Liu Y, Li X, Wang K, Yuan H. Improving biodegradability and biogas production of corn Stover through sodium hydroxide solid state pretreatment. Energy Fuel. 2008;22(4):2761–6.
Zhao T, Li H. Study on ruminal degradation of mainly protein and fiber sources in dairy diets. Contemp Anim Husb. 2009;11:29–32.
Wang B, Mao S, Yang H, Wu Y, Wang J, Li S, Shen Z, Liu J. Effects of alfalfa and cereal straw as a forage source on nutrient digestibility and lactation performance in lactating dairy cows. J Dairy Sci. 2014;97(12):7706–15.
Sun HZ, Wang DM, Wang B, Wang JK, Liu HY, Guan le L, Liu JX. Metabolomics of four biofluids from dairy cows: potential biomarkers for milk production and quality. J Proteome Res. 2015;14(2):1287–98.
Wang D, Liang G, Wang B, Sun H, Liu J, Guan LL. Systematic microRNAome profiling reveals the roles of microRNAs in milk protein metabolism and quality: insights on low-quality forage utilization. Sci Rep. 2016;6:21194.
Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 2011;12(2):87–98.
Choi I, Bao H, Kommadath A, Hosseini A, Sun X, Meng Y, Stothard P, Plastow GS, Tuggle CK, Reecy JM, et al. Increasing gene discovery and coverage using RNA-seq of globin RNA reduced porcine blood samples. BMC Genomics. 2014;15:954.
Ghazalpour A, Bennett B, Petyuk VA, Orozco L, Hagopian R, Mungrue IN, Farber CR, Sinsheimer J, Kang HM, Furlotte N, et al. Comparative analysis of proteome and transcriptome variation in mouse. PLoS Genet. 2011;7(6):e1001393.
Jiang X, Zeng T, Zhang S, Zhang Y. Comparative proteomic and bioinformatic analysis of the effects of a high-grain diet on the hepatic metabolism in lactating dairy goats. PLoS One. 2013;8(11):e80698.
Huang S, Chen L, Te R, Qiao J, Wang J, Zhang W. Complementary iTRAQ proteomics and RNA-seq transcriptomics reveal multiple levels of regulation in response to nitrogen starvation in Synechocystis sp. PCC 6803. Mol BioSyst. 2013;9(10):2565–74.
Nagaraj N, Wisniewski JR, Geiger T, Cox J, Kircher M, Kelso J, Paabo S, Mann M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol Sys Biol011. 7:548.
Greco TM, Cristea IM. Proteomics tracing the footsteps of infectious disease. Mol Cell Proteomics. 2017;16(4 suppl 1):S5–S14.
Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M, Lightfoot S, Menzel W, Granzow M, Ragg T. The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol. 2006;7:3.
Li Y, Lin B, Yang L. Comparative transcriptomic analysis of multiple cardiovascular fates from embryonic stem cells predicts novel regulators in human cardiogenesis. Sci Rep. 2015;5:9758.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc. 2012;7(3):562–78.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25(4):402–8.
Meng Q, Hou L, Zhao Y, Huang X, Huang Y, Xia S, Gu W, Wang W. iTRAQ-based proteomic study of the effects of Spiroplasma eriocheiris on Chinese mitten crab Eriocheir sinensis hemocytes. Fish Shellfish Immunol. 2014;40(1):182–9.
Kong Q, Tong Q, Lou D, Ding J, Zheng B, Chen R, Zhu X, Chen X, Dong K, Lu S. Quantitative proteomic analyses of Schistosoma japonicum in response to artesunate. Mol BioSyst. 2015;11(5):1400–9.
Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;1(72):248–54.
Yang Y, Zheng N, Zhao X, Zhang Y, Han R, Ma L, Zhao S, Li S, Guo T, Wang J. Proteomic characterization and comparison of mammalian milk fat globule proteomes by iTRAQ analysis. J Proteome. 2015;116:34–43.
Sheng Q, Dai J, Wu Y, Tang H, Zeng R. BuildSummary: using a group-based approach to improve the sensitivity of peptide/protein identification in shotgun proteomics. J Proteome Res. 2012;11(3):1494–502.
Zhu W, Fu Y, Wang B, Wang C, Ye JA, Wu YM, Liu JX. Effects of dietary forage sources on rumen microbial protein synthesis and milk performance in early lactating dairy cows. J Dairy Sci. 2013;96(3):1727–34.
Long M, Zhao J, Li T, Tafalla C, Zhang Q, Wang X, Gong X, Shen Z, Li A. Transcriptomic and proteomic analyses of splenic immune mechanisms of rainbow trout (Oncorhynchus mykiss) infected by Aeromonas salmonicida subsp. salmonicida. J Proteome. 2015;122:41–54.
Qiao J, Huang S, Te R, Wang J, Chen L, Zhang W. Integrated proteomic and transcriptomic analysis reveals novel genes and regulatory mechanisms involved in salt stress responses in Synechocystis sp. PCC 6803. Appl Microbiol Biot. 2013;97(18):8253–64.
Camon E, Magrane M, Barrell D, Lee V, Dimmer E, Maslen J, Binns D, Harte N, Lopez R, Apweiler R. The gene ontology annotation (goa) database: sharing knowledge in uniprot with gene ontology. Nucleic Acids Res. 2004;32(suppl 1):D262–6.
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36(Database issue):D480–4.
Wool IG. The structure and function of eukaryotic ribosomes. Annu Rev Biochem. 1979;48(1):719–54.
Wang B, Sun HZ, Xu NN, Zhu KJ, Liu JX. Amino acid utilization of lactating dairy cows when diets are changed from an alfalfa-based diet to cereal straw-based diets. Anim Feed Sci Tech. 2016;217:56–66.
Turashvili G, Bouchal J, Burkadze G, Kolar Z. Wnt signaling pathway in mammary gland development and carcinogenesis. Pathobiology. 2006;73(5):213–23.
Valcourt U, Kowanetz M, Niimi H, Heldin CH, Moustakas A. TGF-beta and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol Biol Cell. 2005;16(4):1987–2002.
Bailey JP, Nieport KM, Herbst MP, Srivastava S, Serra RA, Horseman ND. Prolactin and transforming growth factor-beta signaling exert opposing effects on mammary gland morphogenesis, involution, and the Akt-forkhead pathway. Mol Endocrinol. 2004;18(5):1171–84.
McManaman JL, Neville MC. Mammary physiology and milk secretion. Adv Drug Deliver Rev. 2003;55(5):629–41.
Nath S, Villadsen J. Oxidative phosphorylation revisited. Biotechnol Bioeng. 2015;112(3):429–37.
Holtenius P, Holtenius K. New aspects of ketone bodies in energy metabolism of dairy cows: a review. Zentralblatt fur Veterinarmedizin Reihe A. 1996;43(10):579–87.
Varnam GC, Jeacock MK, Shepherd DA. Hepatic ketone-body metabolism in developing sheep and pregnant ewes. Brit J Nutr. 1978;40(2):359–67.
Graber M, Kohler S, Kaufmann T, Doherr MG, Bruckmaier RM, van Dorland HA. A field study on characteristics and diversity of gene expression in the liver of dairy cows during the transition period. J Dairy Sci. 2010;93(11):5200–15.
Shennan D, Boyd C. The functional and molecular entities underlying amino acid and peptide transport by the mammary gland under different physiological and pathological conditions. J Mammary Gland Biol. 2014;19(1):19–33.
Wang B, Wu G, Zhou Z, Dai Z, Sun Y, Ji Y, Li W, Wang W, Liu C, Han F, et al. Glutamine and intestinal barrier function. Amino Acids. 2014:1–12.
Ferreira-Cerca S, Poll G, Gleizes PE, Tschochner H, Milkereit P. Roles of eukaryotic ribosomal proteins in maturation and transport of pre-18S rRNA and ribosome function. Mol Cell. 2005;20(2):263–75.
Krieg UC, Johnson AE, Walter P. Protein translocation across the endoplasmic reticulum membrane: identification by photocross-linking of a 39-kD integral membrane glycoprotein as part of a putative translocation tunnel. J Cell Biol. 1989;109(5):2033–43.
Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperone-mediated protein folding in the cytosol. Nat Rev Mol Cell Biol. 2004;5(10):781–91.
Vos MJ, Hageman J, Carra S, Kampinga HH. Structural and functional diversities between members of the human HSPB, HSPH, HSPA, and DNAJ chaperone families. Biochemistry. 2008;47(27):7001–11.
Wang WA, Groenendyk J, Michalak M. Endoplasmic reticulum stress associated responses in cancer. BBA-Mol Cell Res. 2014;1843(10):2143–9.
Furuya M, Funasaki M, Tani H, Sasai K. Identification of novel tumour-associated antigens in canine mammary gland tumour. Vet Comp Oncol. 2015;13(3):194–202.
Yasuda K, Nakai A, Hatayama T, Nagata K. Cloning and expression of murine high molecular mass heat shock proteins, HSP105. J Biol Chem. 1995;270(50):29718–23.
Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem. 1999;68(1):1015–68.
Ciechanover A, Orian A, Schwartz AL. Ubiquitin-mediated proteolysis: biological regulation via destruction. Bioessays. 2000;22(5):442–51.
Dye BT, Schulman BA. Structural mechanisms underlying posttranslational modification by ubiquitin-like proteins. Annu Rev Biophys Biomol Struct. 2007;36:131–50.
Van Wijk SJ, Timmers HM. The family of ubiquitin-conjugating enzymes (E2s): deciding between life and death of proteins. FASEB J. 2010;24(4):981–93.
Seedorf U, Ellinghaus P, Nofer JR. Sterol carrier protein-2. BBA-Mol Cell Biol L. 2000;1486(1):45–54.
Cherbavaz DB, Lee ME, Stroud RM, Koshland Jr DE. Active site water molecules revealed in the 2.1 Å resolution structure of a site-directed mutant of isocitrate dehydrogenase1. J Mol Biol. 2000;295(3):377–85.
Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, Thompson CB. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. P Nat Acad Sci USA. 2011;108(49):19611–6.
Thorpe C, Kim J. Structure and mechanism of action of the acyl-CoA dehydrogenases. FASEB J. 1995;9(9):718–25.
Acknowledgements
The authors gratefully thank the personnel of Hangjiang Dairy Farm (Hangzhou, China) for their assistance in the feeding and care of the animals.
Funding
This research was supported by grants from the National Key Research and Development Program of China (2016YFD0500503) and the National Natural Science Foundations of China (31672447, 31372336). The funders had no role in study design, data collection, analysis and interpretation, or preparation of the manuscript.
Availability of data and materials
All data in this study are included in the article and its additional files.
Author information
Authors and Affiliations
Contributions
DWT and LHY conceived of the study and designed the project. DWT performed the lab work, analyzed data and wrote the manuscript. WQJ helped the qRT-PCR experiment and the KEGG pathway analysis. LHY, ZFQ and LJX revised the manuscript. All the authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The procedures of this study were approved by the Animal Care and Use Committee of Zhejiang University (Hangzhou, China) and were in accordance with the university’s guidelines for animal research.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Primers used in real-time RT-PCR. (XLSX 11 kb)
Additional file 2:
Table S2. Summary of the transcriptome data in the mammary gland of dairy cows fed corn stover (CS) or alfalfa hay (AH). Table S3. Summary of the proteome data in the mammary gland of dairy cows fed corn stover (CS) or alfalfa hay (AH)). (ZIP 13 kb)
Additional file 3:
Table S4. Differentially expressed genes in the mammary gland of cows fed corn stover (CS) vs. alfalfa hay (AH). The cutoff is set at 1.5-fold change and p < 0.05. Table S5. Differentially expressed proteins in the mammary gland of cows fed corn stover (CS) vs. alfalfa hay (AH). The cutoff is set at 1.2-fold change and p < 0.05. Table S6. Differentially expressed genes found in both transcriptomic and proteomic analyses in the mammary gland of cows fed corn stover (CS) vs. alfalfa hay (AH). (ZIP 164 kb)
Additional file 4:
Table S7. A list of common differentially expressed genes and differentially expressed proteins in the mammary gland of cows fed corn stover (CS) vs. alfalfa hay (AH). (XLSX 17 kb)
Additional file 6:
Table S9. The full name and abbreviation of proteins in Fig. 8. (XLSX 16 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Dai, W., Wang, Q., Zhao, F. et al. Understanding the regulatory mechanisms of milk production using integrative transcriptomic and proteomic analyses: improving inefficient utilization of crop by-products as forage in dairy industry. BMC Genomics 19, 403 (2018). https://doi.org/10.1186/s12864-018-4808-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-4808-5