
Abstract
Background
Microdeletion of chromosome 22q11 is associated with significant developmental anomalies, including disruption of the cardiac outflow tract, thymic/parathyroid aplasia and cleft palate. Amongst the genes within this region, TBX1 is a major candidate for many of these developmental defects. Targeted deletion of Tbx1 in the mouse has provided significant insight into the function of this transcription factor during early development of the cardiac and pharyngeal systems. However, less is known about its role during palatogenesis. To assess the influence of Tbx1 function on gene expression profile within the developing palate we performed a microarray screen using total RNA isolated from the secondary palate of E13.5 mouse embryos wild type, heterozygous and mutant for Tbx1.
Results
Expression-level filtering and statistical analysis revealed a total of 577 genes differentially expressed across genotypes. Data were clustered into 3 groups based on comparison between genotypes. Group A was composed of differentially expressed genes in mutant compared to wild type (n = 89); Group B included differentially expressed genes in heterozygous compared to wild type (n = 400) and Group C included differentially expressed genes in mutant compared to heterozygous (n = 88). High-throughput quantitative real-time PCR (RT-PCR) confirmed a total of 27 genes significantly changed between wild type and mutant; and 27 genes between heterozygote and mutant. Amongst these, the majority were present in both groups A and C (26 genes). Associations existed with hypertrophic cardiomyopathy, cardiac muscle contraction, dilated cardiomyopathy, focal adhesion, tight junction and calcium signalling pathways. No significant differences in gene expression were found between wild type and heterozygous palatal shelves.
Conclusions
Significant differences in gene expression profile within the secondary palate of wild type and mutant embryos is consistent with a primary role for Tbx1 during palatogenesis.
Similar content being viewed by others


Background
22q11.2 deletion syndrome (22q11.2DS) is the most common human microdeletion [1] occurring with a prevalence of 1:4000 and incidence ranging from 1:2000–6395 [2,3,4]. This microdeletion is associated with several syndromic conditions including DiGeorge (DGS; MIM 188400), velocardiofacial (VCFS; MIM 192430), conotruncal anomaly face (CAFS or Takao syndrome; MIM 217095) and isolated outflow tract (OFT) defects of the heart [5,6,7,8,9]. These conditions are characterized predominantly by the presence of congenital heart defects, thymic and parathyroid hypoplasia, and craniofacial dysmorphism, including oro-facial clefting that predominates as isolated cleft palate, micrognathia and (less commonly) dental defects [10,11,12,13]. The most common deletions are phenotypically indistinguishable from each other and consist of either a 3 Mb segment spanning the low copy repeats (LCR) A-D (around 85% of cases); or a smaller 1.5 Mb deletion that spans LCR A-B seen in around 15% of cases [14,15,16]. A less common LCR C-D deletion of the typical 22q11.2DS region has also been identified, which is associated with a much-reduced prevalence of cardiac malformations and oro-facial clefting [17,18,19]. 22q11.2DS is a contiguous gene and haploinsufficient syndrome with at least 30 different genes potentially contributing to the characteristic clinical features [20, 21]. Amongst the genes identified as candidates for the development of 22q11.2DS, T-Box 1 (TBX1), which encodes a T-Box-containing transcription factor is recognised as a major determinant through its location within the 22q11 critical region [21,22,23], expression in organs affected within the clinical spectrum [24,25,26,27] and observations that loss of Tbx1 function in mouse recapitulates the clinical findings seen in many DGS subjects [23, 28,29,30,31]. Supporting this, TBX1 mutation has been identified in a sporadic case of DGS [32] and Tbx1 haploinsufficiency results in the most characteristic phenotypes related to developmental defects in the embryonic pharyngeal apparatus [32, 33]. DGS is also referred to as the III-IV pharyngeal pouch syndrome, as the pharyngeal pouches and their associated blood vessels are the structures most commonly affected [23, 30]. Apart from the aortic arch, thymus and parathyroid gland defects, Tbx1 murine models also manifest craniofacial anomalies that arise from developmental defects associated with pharyngeal arches I and II [23, 34, 35]. Indeed, conditional mutant models have revealed a tissue-specific requirement and a dose sensitivity for Tbx1 during murine pharyngeal development [20, 36,37,38].
The majority of 22q11.2DS individuals have a characteristic craniofacial morphology including lateral displacement of the inner canthi, swollen eyelids, small mouth, hypoplastic mandible, flat nasal bridge and square nose [39,40,41]. Cleft palate (including submucous cleft) is also present in approximately 10% of subjects [40]. Morphological studies to assess embryonic malformations in various Tbx1 genotypes also reveal the presence of cleft palate in Tbx1-overexpressing mice [42, 43]. Therefore, both loss and gain of Tbx1 function can lead to the development of a cleft phenotype.
The palate is divided anatomically into primary and secondary regions with the secondary palate composed of both hard and soft tissues. Embryologically, the secondary palate is derived from the paired maxillary processes of pharyngeal arch I, which gives rise to the palatal shelves. During palatogenesis, these shelves are initially situated bilaterally adjacent to the developing tongue; however, progressive growth and elevation results in them positioning themselves above the tongue, with further medial growth leading to fusion with their counterpart along the midline to create a single continuous palate. The palatal shelves also fuse with the nasal septum superiorly and primary palate anteriorly, completing separation of the nasal and oral cavities [44,45,46]. In the developing mouse embryo, Tbx1 is expressed in epithelium of the palatal shelves throughout palatogenesis from embryonic day (E)12.5–15.5 [24]. The etiological basis of the cleft palate phenotype in Tbx1 mutants is not fully understood but has been associated with abnormal palatal shelf elevation, possibly due to a combination of increased tongue height, decreased palatal shelf width, perturbed cell proliferation and apoptosis [47]. In addition, inappropriate fusion between the palatal shelf epithelium and tongue has also been described in this mutant, associated with hyper-proliferation and disrupted differentiation [48]. More recently, confocal image analysis has found only subtle differences in levels of proliferation within mesenchyme of the palatal shelves between wild-type and mutant until the later stages of palatogenesis; although significant differences in mesenchymal cell orientation were found in mutant shelves, which might contribute to the cleft phenotype [49].
We are interested in further defining the role of Tbx1 during the process of murine palatogenesis. Specifically, we have investigated regulation of this transcription factor in the secondary palate and carried out a functionally-based microarray using the Tbx1 mouse model. We compared total RNA isolated from dissected secondary palatal shelves derived from E13.5 wild type (WT), Tbx1+/− (heterozygous) and Tbx1−/− (mutant) embryos and clustered the data into three groups based on comparison between the three genotypes. Microarray analysis demonstrated that in the absence of functional Tbx1, significant changes occur in the expression profile of numerous genes in mutant versus WT and mutant versus heterozygous groups. The most significant pathways affected in both groups were the hypertrophic cardiomyopathy, cardiac muscle contraction, dilated cardiomyopathy, focal adhesion, calcium signalling and tight junction pathways. High-throughput quantitative RT-PCR validation confirmed significant variation between WT and mutant in the expression of 26 individual genes. We discuss these findings within the context of murine secondary palatogenesis.
Results
Regulation of Tbx1 in the developing secondary palate
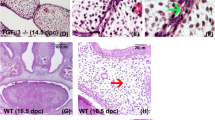
Tbx1 transcriptional activity is present in epithelium of the secondary palate shelves throughout the processes of growth, elevation and fusion (Additional file 1) and Tbx1 mutant mice have a fully penetrant cleft palate [23, 30, 31]. We are interested in further defining the function of this transcription factor during palatogenesis at the molecular level and first sought to understand how Tbx1 transcription might be regulated in the palatal shelf epithelium. We began by investigating the effect of abrogating either Sonic hedgehog (Shh) or Fibroblast growth factor (Fgf) signaling in palatal shelf explants as there are potential associations between these signaling networks and Tbx1 function in the developing palate. Shh is also expressed in the palatal epithelium and lies upstream of Tbx1 in the pharyngeal endoderm [50]; whilst Fgf signaling can maintain Tbx1 expression in early odontogenic epithelium [27]. Specifically, E13.5 secondary palatal shelves were isolated and cultured for 24 h in the presence of either the Shh antagonist cyclopamine or the Fgf receptor inhibitor SU4502. Interestingly, whilst an absence of Shh signaling did not affect Tbx1 transcription, loss of Fgf signaling resulted in a loss of Tbx1 activity in the palatal epithelium after 24 h of culture (Fig. 1a-g). These results place Tbx1 downstream of Fgf signaling during early palatogenesis and in contrast to the pharyngeal region, loss of Shh does not affect Tbx1.

Regulation of Tbx1 expression in the early secondary palate. Wholemount in situ hybridization on palatal shelf explants cultured for 24 h in the presence or absence of the Shh inhibitor cyclopamine and the Fgf receptor inhibitor SU5402. a Tbx1 is expressed in the palatal shelf epithelium and first molar tooth germ (arrowed); (b) in the absence of Shh signaling, Tbx1 is maintained; (c) in the absence of Fgf signaling, Tbx1 is lost; (d) Shh signaling is active in the developing palate and first molar (arrowed) as shown by expression of the Shh transcriptional target patched1 (Ptch1); (e) in the presence of cyclopamine Ptch1 transcription is lost; (f) Fgf signaling is active in the developing palate and first molar (arrowed), as shown by expression of the Fgf transcriptional target sprouty2 (Spry2); (g) in the presence of SU4502 Spry2 is lost. Lines mark the medial edge of the palatal shelf
Altered gene expression in the secondary palate of Tbx1 mutant mice
It is known that Shh, Fgf and Bone morphogenetic protein (Bmp) signaling pathways are important during normal development of the palate [51,52,53]; in particular, reciprocal signaling between epithelial Shh and mesenchymal Fgf10, mediated through fibroblast growth factor receptor 2b (Fgfr2b), regulates cell proliferation in the mesenchyme [54]. Whilst Shh also negatively regulates Bmp4 in the mesenchyme, which is itself upstream of Fgf10 [55]. Tbx1 interacts with a number of these molecules during embryogenesis, being directly upstream of Fgf10 in the early heart field [28, 56]; negatively modulating Bmp4 through the binding of Smad1 in cardiomyocytes [36] and being downstream of Shh in endoderm of the early pharynx [50]. Within the palate itself, it has been variously suggested that Tbx1 negatively regulates Fgf10 and Bmp4, whilst positively regulating Fgf8 and Pax9, although there is currently not a consensus on these findings [47, 48].
Although we could find no evidence that Tbx1 is downstream of Shh signaling in the palatal epithelium, there is considerable overlap of expression. We therefore investigated known targets of Shh within palatal shelves WT and mutant for Tbx1 using in situ hybridization. Interestingly, we found no significant differences in expression of Shh, Fgf10 and Fgfr2b between WT and mutant (Fig. 2a-f). However, whilst Fgf8 expression was also normal in the mutant shelves (Fig. 2g-h), Bmp4 and paired-box 9 (Pax9) were slightly up and downregulated, respectively in the posterior region of the secondary palate (Fig. 2i-l). These apparent changes in Bmp4 and Pax9 expression in the mutant might simply be a function of altered numbers of cells expressing these genes in the palate mesenchyme, particularly as the Tbx1 domain within the palatal epithelium does not completely overlie those of Bmp4 or Pax9 in the mesenchyme [48]. However, given the evidence of retarded growth in Tbx1 mutant palatal shelves [47, 48] if an alteration in cell number is responsible for any of these changes, it would seem to be more likely for Pax9.

Signaling interactions during development of the secondary palate in WT and Tbx1 mutant embryos. Section in situ hybridization demonstrating the expression of key signaling molecules. a, b Shh; (c, d) Fgf10; (e, f) Fgfr2b; (g, h) Fgf8; (i, j) Bmp4; (k, l) Pax9
Microarray analysis
To further identify potential transcriptional target genes of Tbx1 implicated in palatogenesis, microarray analysis was carried out using cDNA transcribed from total RNA derived from the dissected secondary palatal shelves of E13.5 Tbx1+/+; Tbx1+/− and Tbx1−/− embryos (n = 3 for each genotype).
After normalization and filtering of microarray data, comparison between mutant embryos and WT (Group A), heterozygous and WT (Group B) and mutant versus heterozygous (Group C) were performed (adj. p < 0.1). The WebGestalt database was used to identify biological pathways associated with these differentially expressed transcripts [57]. In Group A, 89 genes were identified to be differentially expressed in mutant compared to WT (adj. p < 0.1, fold change 1.4). From these, 3 genes were upregulated, whereas the majority (n = 86) were downregulated (Table 1). Group B includes differentially expressed genes arising from the comparison of heterozygous and WT palates (n = 400, adj. p > 0.23). This group list was not considered statistically significant (adj. p > 0.1) and therefore was not analysed further (Additional file 2). In Group C, 88 genes were identified to be differentially expressed in the mutant compared to heterozygote palate (adj. p < 0.1, fold change 1.3). Amongst these, 11 genes were upregulated, whereas 77 were downregulated (Table 1). In Group A, from the 89 genes that were searched, 9 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were identified (Fig. 3a). The most statistically enriched pathways (adj. p < 0.1) were all associated with cardiac muscle physiology and included hypertrophic cardiomyopathy, cardiac muscle contraction, dilated cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy and vascular smooth muscle contraction. Other pathways included phagosome and focal adhesion, tight junction and calcium signaling pathways and Alzheimer's disease (Additional file 3). In Group C, from the 88 genes that were searched, 10 KEGG pathways were identified (Fig. 3b). The most statistically enriched pathways (adj. p < 0.1) were all also associated with cardiac muscle physiology, including hypertrophic and dilated cardiomyopathy and arrhythmogenic right ventricular cardiomyopathy. Other pathways included tight junction, calcium signalling, focal adhesion, neuroactive ligand-receptor interaction, phagosome and Alzheimer’s disease pathways (Additional file 3). We were then interested to further identify the proportion of overlap amongst significantly differentially expressed genes between Groups A and C. (Fig. 4a [58]). The two groups share 58 commonly expressed genes (Table 2) when compared to WT and heterozygous; whereas 30 genes (Table 2) were uniquely observed in Group A and 20 in Group C (Table 2; adj. P < 0.1). The WebGestalt database was used to provide insights into the mechanism of regulation associated with these 58 common gene transcripts. Adding to the above approach, heat map and dendogram clustering of the commonly expressed genes, as well as uniquely expressed genes in Group A and Group C (n = 99 genes) revealed transcriptional homogenicity between genotypes (Fig. 4b). Genes upregulated in mutants clearly clustered together and were shown to be downregulated in heterozygote and WT samples (red asterisks in Fig. 4b). In contrast, the downregulated transcriptome of mutant samples was shown to increase its expression in heterozygous and WT palates. Although statistical analysis revealed a non-significant expression pattern of Tbx1 heterozygous samples (adj. p values > 0.1), heat map revealed a similarity in gene expression pattern between heterozygous and WT samples.

a Pathway analysis of genes differentially expressed in the Tbx1 mutant secondary palate compared to WT (Group A); (b) pathway analysis of genes differentially expressed in the mutant secondary palate when compared to heterozygous (Group C): The pie chart depicts the number of assigned genes for each significantly enriched pathway. Data sets are illustrated as slices, the sizes of which are proportional to the number of genes implicated in each pathway. The ten pathways are listed and colour-coded on the right

a Pairwise Venn diagram illustrating the comparison between gene sets from Tbx1 mutant secondary palate compared to WT (Group A) and Tbx1 mutant compared to heterozygous (Group C). The Venn diagram identified 58 common elements between Group A and Group C. Numbers in each section represent the number of genes. Transcripts utilized for the construction of the Venn diagram were statistically significant with adj. p values < 0.1; (b) heat map (hierarchical clustering) of commonly expressed genes in Groups A and C, as well as uniquely expressed genes in Group A and C. Hierarchical cluster of 99 genes found to be differentially expressed in the 3 mutant, 3 heterozygous and 3 WT palatal samples. Transcripts utilized for the construction of clustering were statistically significant with adj. p values < 0.1 except for heterozygous where adj. p values were > 0.1. Visual inspection of heat map and dendogram clustering of the 9 samples revealed that all triplicates of the same genotype clustered together. Upregulated genes in mutants clustered together (red asterisks on left) and their pattern of expression could be visibly compared top heterozygous and WT samples. Each row represents a specific gene, and each column represents each genotype of the samples analysed. The colour represents the expression level of the gene. Red represents high expression, while blue represents low expression. The expression levels are continuously mapped on the colour scale provided at the top left of the figure. The dendrogram at the top of the matrix provides the degree of similarity between examined groups assessing the similarity between expressed genes and samples used for comparison. Note the similarity in gene expression between WT and Tbx1 heterozygous transcripts
Confirmation of microarray data
For validation of the results obtained by microarray, RT-PCR was carried out using gene-specific primers (Applied Biosystems; Additional file 4) and the original RNA samples. In total, 27 genes from Group A and 28 genes from Group C were selected for gene expression verification (Table 3). Changes in gene expression of these transcripts were normalized to that of ß-Actin. In both groups, 27 genes were commonly expressed (Table 3; Fig. 5a); Alas2 was uniquely present in Group A, whereas Ank1 and Chrna1 were uniquely present in Group C (Table 3; Fig. 5b). All genes tested were confirmed as being significantly changed between WT-mutant and heterozygote-mutant except for Ank1 (Group C; p = 0.102). In Group A, Rapsn, Sh3bgr, Tnnc2, Tnni2 and Tnnt2 were the most downregulated genes; whereas in Group C, these were Csrp3, Sh3bgr, Sln, Tnnc2, Tnni2, Myh7 and Mylpf.

Quantitative reverse transcriptase polymerase chain reaction verification of genes identified in Groups A and C following the microarray analysis. a Common genes significantly changed between both WT-mutant and heterozygote-mutant; (b) genes significantly changed only between WT-mutant (Group A); (c) genes significantly changed only between heterozygote-mutant (Group C)
Discussion
In the present study, functional explant assays and microarray analysis of gene expression was carried out in the palatal shelves of E13.5 mouse embryos WT, heterozygous or mutant for Tbx1. This was prompted by the knowledge that Tbx1 is strongly expressed in epithelium of the palatal shelves throughout palatogenesis, mutant embryos demonstrate cleft palate with complete penetrance [23, 24, 47, 48] and the findings that Tbx1 has multiple potential roles during normal palatal shelf elevation, elongation and adhesion [47, 48]. It is known that several regulatory networks underlie signaling between epithelium and mesenchyme during development of the secondary palate and we sought to discover potential genetic pathways disrupted during palatogenesis in the absence of Tbx1. We therefore focused our investigations at E13.5, just prior to the period of rapid growth and elevation [45].
A key finding of this profile is the association between an absence of Tbx1 function and altered expression (primarily downregulation) in a number of muscle-related genes within the shelves of the secondary palate. Developing mononuclear and binucleate myofibril-containing skeletal muscle cells are identifiable within the palatal shelves at E13 [59] and findings of altered gene expression are perhaps not surprising, given the essential role of Tbx1 during the development of branchiomeric musculature and somite-derived tongue muscles [60,61,62] and detectable expression in adult mouse muscle [63, 64]. In the embryo, Tbx1 activates the myogenic-determination genes myogenic factor 5 (Myf5) and myogenic differentiation (MyoD) in the mesodermal core of pharyngeal arches I and II [61]. In addition, loss of Tbx1 results in impairment of the onset of myogenic specification [60] and Tbx1 synergizes with the myogenic factor Myf5 for initiation of myogenic cell fate [65]. Our array failed to identify variation in Myf5 and MyoD, but verified downregulation of Myf7 at E13.5 in mutant palatal shelves. This finding suggests that Tbx1 functions upstream of myosin heavy chain 7 (Myh7) during palatal shelf formation and just prior to elevation, possibly as a myogenic factor. The presence of asymmetric expression patterns of myogenic regulatory factors in early first arch-derived muscles of Tbx1 mutant embryos might explain the absence of Myf5 and MyoD gene transcripts [61]. In addition, both skeletal, smooth and non-muscle contractile systems have been identified and implicated in the process of normal palatal shelf elevation [66, 67]. A number of the downregulated genes identified have also been implicated in the process of skeletal and cardiac muscle contraction (Tnni2, Tnnt1, Myh3, Myom1, Tnnc2), which might reflect the lack of skeletal myogenic determination. Interestingly, microarray analysis of the early pharyngeal region of Df1/+; Tbx1+/− embryos has previously demonstrated upregulation of Tnnc2 [68]. It cannot be discounted that other intrinsic contractile systems might also be disrupted in the secondary palate of Tbx1 mutant mice. Indeed, changes in expression levels were also identified in genes associated with intracellular calcium signaling (Atp2a1, Tnnc2, Cacna1s, Tnnc1), which is known to mediate a number of important physiological processes of relevance to palatogenesis, including skeletal and smooth muscle contraction, apoptosis, cell motility and proliferation [69].
After palatal shelf elevation, periderm cells joined by tight junctions are believed to function as a protective layer, preventing aberrant adhesions and playing an important role in mediating appropriate shelf adherence and epithelial differentiation [70, 71]. Loss of periderm is required at the tips of opposing palatal shelves and overall at sites where fusion is required [71]. In Tbx1 mutant mice, aberrant oral adhesions between tongue and palatal shelves have been observed [48]. In the present study, the tight junction genes Myh3, Mylpf, Myh7 and Actn2 were downregulated in mutants at E13.5, suggesting a potential role for Tbx1 in the normal function of tight junctions present within the palatal shelf epithelium.
Comparison between WT-mutant and heterozygous-mutant shelves revealed 58 genes commonly expressed in both groups. From these, 27 genes from Group A and 28 genes from Group C were selected for gene expression verification. Analysis revealed significant downregulation of 26 genes common to both groups (see Fig. 5a) with (Alas2) and (Ank1, Chrna1) individually downregulated in each group, respectively (see Fig. 5b). Statistical analysis revealed significant downregulation of all genes tested through RT-PCR with the exception of Ank1 (p = 0.102; see Fig. 5b). Pathway analysis of these validated genes confirmed the associations between cardiac muscle contraction and calcium signaling, but also suggested links with dilated and hypertrophic cardiomyopathies. Although 22q11.2DS is commonly associated with conotruncal congenital heart defects, hypocalcemic dilated myocardiopathy has also been described in association with this condition [72]. RT-PCR validation of the microarray analysis demonstrated no significant changes in gene expression between WT and heterozygous shelves, consistent with the normal palatogenesis seen in heterozygous embryos [23].
Tbx1 is known to regulate both Fgf8 and Fgf10 expression in the early pharyngeal arches and cardiac outflow tract [64] and influence the spatial distribution of Fgf8 and Bmp4 in the early mandible [73]. It has also been suggested that Fgf8 is significantly downregulated in the palatal shelf epithelium, whilst Fgf10 is upregulated in the mesenchyme at E13.5 in Tbx1 mutant embryos [47]. However, we found no evidence of altered transcript levels associated with these genes in our array. This same report also demonstrated diminished hyaluronic acid (HA) in the palatal shelves of Tbx1 mutant mice and whilst we found no obvious genetic links to this finding within our array, HA has been shown to induce matrix metalloproteinase 9 (MMP9) [74], which was downregulated. However, whilst some members of the MMP family have been directly related to palatogenesis, at least in vitro; this did not include MMP9 [75].
In this microarray experiment, RNA was derived from whole dissected palatal shelves and therefore no formal distinction was made between changes in epithelial and mesenchymal gene activity. Tbx1 is localized to the palatal shelf epithelium at E13.5, but is clearly able to influence signaling activity between epithelium and mesenchyme in the palate (Fig. 6). Indeed, the associations between Tbx1 function and muscle contraction and calcium signaling, both activities that take place in the early mesenchyme, are consistent with this. In addition, Tbx1 seems to act co-operatively with Shh signaling in the palate, through the repression of Bmp4 and induction of Pax9. Interestingly, this co-operative activity would appear to be dependent upon Fgf signaling; Shh in the epithelium is dependent upon reciprocal signaling with Fgf10 in the mesenchyme [54] and our explant studies demonstrate that Tbx1 is also dependent upon Fgf signaling. Although it is currently not known which Fgf ligand is required or whether this is within the epithelium or mesenchyme, maintenance of epithelial Tbx1 transcription is essential for normal palatogenesis. Conditional loss of Tbx1 in either craniofacial mesenchyme [48] or mesoderm [76] does not result in cleft palate, in contrast to loss-of-function in the oral epithelium, which does [48].

Molecular associations linking Tbx1 with Fgf and Shh signaling in the developing palate. Tbx1 in the palatal shelf epithelium is downstream of Fgf signaling, the ligand/s and source (epithelium/ mesenchyme) are currently unknown. Shh-Fgf10-Fgfr2b epithelial-mesenchymal reciprocal signaling [54] antagonizes Bmp4 [55] and induces Pax9 indirectly through the induction of Osr2 [55, 82]. We and others [48] have demonstrated that Tbx1 acts to inhibit Bmp4 and induce Pax9. It has been suggested that Tbx1 activity is required for Fgf8 induction in the epithelium and Fgf10 inhibition in the mesenchyme [47]; however, we and others [48] have found no evidence of this
Conclusions
We have conducted functional microarray analysis and PCR validation of gene expression in the developing secondary palate at E13.5 in the Tbx1 mutant embryo. Differentially regulated genes were detected in the absence of this transcription factor. In the microarray, a total of 89 genes demonstrated differential expression in Group A and 88 genes in Group C (adj. p < 0.1), whilst high-throughput quantitative RT-PCR confirmed 27 genes significantly changed between WT and mutant and 28 between heterozygote and mutant. Associations existed with cardiac muscle development, hypertrophic and dilated cardiomyopathy, tight junction and calcium signaling. These findings provide further evidence of a primary role for Tbx1 during the process of palatogenesis.
Methods
Mice
Breeding mice were maintained in ventilated cages on an alternating (12:12) light-dark cycle in the Biological Services Unit at King’s College London. Time-mated Tbx1 embryos were generated by inter-crossing Tbx1+/− mice on a C57/Bl6 background [23] such that noon of the day on which vaginal plugs were detected was considered as embryonic day (E) 0.5. Pregnant females were euthanized with cervical dislocation.
Explant culture
Secondary palatal shelves were carefully micro-dissected from E13.5 WT embryos and cultured for 24 h in the presence of cyclopamine or SU4502 as previously described [77]. Briefly, explants were cultured using a modified Trowell technique at 37 °C in an atmosphere of 5% CO2 in serum-free Advanced DMEM/F12 (GibcoBRL) supplemented with 20 U/ml penicillin and streptomycin (GibcoBRL), 10% Fetal Bovine Serum (GibcoBRL), 50 mM transferrin (Sigma) and 150 μg/ml ascorbic acid (Sigma). SU5402 (Calbiochem) was diluted in medium from a 10 mM stock solution in DMSO and cyclopamine (Sigma) was diluted from a 20 mg/ml stock solution in ethanol and added to the culture medium at a final concentration of 75 μM for both inhibitors. A minimum of (n = 6) palatal shelves were used for each experiment.
In situ hybridisation
Wholemount digoxygenin and section 35S radioactive in situ hybridisation was carried out as previously described [78]. Wholemount (n = 6 palatal shelves) and section (n = 3 embryos) images were photographed using Leica or Zeiss Axioscop microscopes, respectively. For radioactive in situ hybridisation, light and darkfield images were merged in Adobe photoshop CS. Plasmid cDNA was kindly provided by the following investigators: Bmp4 (Brigid Hogan); Fgf8 (Ivor Mason); Fgf10; Fgfr2b (David Rice); Pax9 (Heiko Peters); Ptch1 (Matthew Scott); Shh (Andy McMahon); Sprty2 (M. Albert Basson), Tbx1 (Peter Scambler).
Tissue preparation and microarray analysis
Secondary palatal shelves were carefully micro-dissected from E13.5 Tbx1 WT, heterozygous or mutant embryos (3 embryos per genotype), stored as pairs from each embryo in RNAlater (Ambion) and then homogenized using a blunt 20-guage needle to an RNase-free syringe. Total RNA was extracted from homogenate derived from each shelf pair using an RNeasy Isolation Kit (Qiagen). RNA quality was checked using an Agilent Bioanalyzer and quantified with spectrophotometry (NanoDrop ND-1000). In total, 9 sets of RNA were collected, each derived from paired secondary palatal shelves harvested from each embryonic genotype (giving 3 samples from each genotype).
Microarray chip processing and data analysis
The expression profiling analysis was carried out at the Franklin-Wilkins Building Genomics Facility, King’s College London. Total RNA was reverse-transcribed and cRNA generated using the MessageAmp II-Biotin Enhanced cRNA Amplification Kit (Ambion). cRNA targets were then hybridized to the Affymetrix Mouse GeneChip microarray (MOE430_A_2 GeneChip array), which is a single array containing 22,690 probe sets representing transcripts and variants from over 14,000 well characterized mouse genes. A single chip was used for each pair of palatal shelves per genotype, with hybridization and scanning of array chips carried out according to recommended protocols (www.affymetrix.com).
Microarray data were analysed by the implementation of Bioconductor packages in the programming language R. Intensity values of every chip were imported and evaluated with the packages affy, simpleaffy and affyPLM. Pre-processing, normalization and expression transformations were executed by the function rma of the affy package [79]. Gene expressions were fitted to linear models and moderated t-statistics were calculated for specific comparisons using lmfit and eBayes functions of the limma package [80]. P-values were adjusted for multiple testing with the Benjamini & Hochberg FDR method [81], implemented within the topTable function of the limma package. Venn diagram and heatmap showing hierarchical clustering with complete linkage scaled by genes were constructed using the packages VennDiagram and gplots respectively. Microarray datasets have been submitted to the Gene Expression Omnibus (GEO) at NCBI (GSE37904).
Functional annotation of differentially regulated gene sets
In this study WEB-based GEne SeT AnaLysis Toolkit (WebGestalt, http://www.webgestalt.org/option.php, version 05/20/2014) was utilized to perform functional enrichment analysis on the data sets containing genes from the Tbx1−/− versus WT shelves comparison (Group A), the Tbx1−/− versus Tbx1+/− shelves comparison (Group C) and the commonly expressed gene set of Group A and Group C. For each gene set, WebGestalt used the hypergeometric test to evaluate functional enrichment against predefined categories collected from KEGG. Statistical analysis was performed according to the current default settings.
Validation with high throughput quantitative real-time RT-PCR and data analysis
Candidate genes were validated with high-throughput real time quantitative RT-PCR using the same nine total RNA samples from the microarray screen. RNA was converted to first-strand cDNA using the High Capacity RNA-to-cDNA kit (Applied Biosystems). Real time PCR assays were identified using Applied Biosystems UmapIt tool to map microarray probeset IDs to inventoried Taqman(r) assays. cDNA samples and assay master mixes were combined on 384-well real-time PCR plates (Applied Biosystems) using the Biomek FX liquid handling robot (Beckman Coulter). A total of nine 384-well plates were used. Each cDNA sample was combined with each gene primer sequence and replicated across four wells, giving four technical replicates for each PCR reaction. Each 384-well plate contained a column for water (no-template control) and ß-Actin (house-keeping gene/endogenous control for data normalization) with a 7900HT Quantitative PCR machine (Applied Biosystems) used for the PCR reaction. The qPCR data was analysed using RQ manager (Applied Biosystems) and Microsoft Excel. The RQ manager uses CT values from the qPCR reaction along with normalisation of the data to provide Relative Quantification (RQ) values (RQ = 2–ΔΔCT) for gene expression. For the 26 commonly expressed genes from Group A and C, ANOVA was used to detect statistically significant differences in Relative Quantification group means between WT, heterozygous and mutant genotypes. The differences in Relative Quantification for the uniquely expressed Alas2 in Group A and Ank1, Chrna1 genes between the WT and the MUT (Group C) were analysed by using t-test. All the above statistical analyses and graphs designs were performed in R. For the graphs, the ggplot2 package was used (see Fig. 5). From the 29 genes selected from microarray analysis, 28 individual genes showed significant changes in expression levels in the mutant compared to WT and/or heterozygote (P value < 0.05), whereas only Agtr2 from Group C was shown to be non-statistically significant (P value = 0.102), in qPCR.
Abbreviations
- 22q11.2DS:
-
22q11.2 deletion syndrome
- Bmp:
-
Bone morphogenetic protein
- CAFS:
-
Conotruncal anomaly face
- DGS:
-
DiGeorge syndrome
- Fgf:
-
Fibroblast growth factor
- Fgfr2b:
-
Fibroblast growth factor receptor 2b
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- Myf5 :
-
Myogenic factor 5
- Myh7:
-
Myosin heavy chain 7
- MyoD :
-
Myogenic differentiation
- OFT:
-
Outflow tract
- Pax9:
-
Paired box 9
- Ptch1:
-
Patched 1
- RT-PCR:
-
Reverse Transcription Polymerase Chain Reaction
- Shh:
-
Sonic hedgehog
- Smad:
-
Mothers against decapentaplegic homologue
- Spry2:
-
Sprouty 2
- TBX1 :
-
Transcription factor-encoding T-Box 1
- VCFS:
-
Velocardiofacial syndrome
- WT:
-
Wild Type
References
Hacihamdioglu B, Hacihamdioglu D, Delil K. 22q11 deletion syndrome: current perspective. Appl Clin Genet. 2015;8:123–32.
Botto LD, May K, Fernhoff PM, Correa A, Coleman K, Rasmussen SA, Merritt RK, O'Leary LA, Wong LY, Elixson EM, et al. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics. 2003;112(1 Pt 1):101–7.
McDonald-McGinn DM, LaRossa D, Goldmuntz E, Sullivan K, Eicher P, Gerdes M, Moss E, Wang P, Solot C, Schultz P, et al. The 22q11.2 deletion: screening, diagnostic workup, and outcome of results; report on 181 patients. Genet Test. 1997;1(2):99–108.
Devriendt K, Fryns JP, Mortier G, van Thienen MN, Keymolen K. The annual incidence of DiGeorge/velocardiofacial syndrome. J Med Genet. 1998;35(9):789–90.
Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet. 2007;370(9596):1443–52.
Scambler PJ, Carey AH, Wyse RK, Roach S, Dumanski JP, Nordenskjold M, Williamson R. Microdeletions within 22q11 associated with sporadic and familial DiGeorge syndrome. Genomics. 1991;10(1):201–6.
Driscoll DA. Genetic basis of DiGeorge and velocardiofacial syndromes. Curr Opin Pediatr. 1994;6(6):702–6.
Scambler PJ, Kelly D, Lindsay E, Williamson R, Goldberg R, Shprintzen R, Wilson DI, Goodship JA, Cross IE, Burn J. Velo-cardio-facial syndrome associated with chromosome 22 deletions encompassing the DiGeorge locus. Lancet. 1992;339(8802):1138–9.
Burn J, Takao A, Wilson D, Cross I, Momma K, Wadey R, Scambler P, Goodship J. Conotruncal anomaly face syndrome is associated with a deletion within chromosome 22q11. J Med Genet. 1993;30(10):822–4.
Klingberg G, Oskarsdottir S, Johannesson EL, Noren JG. Oral manifestations in 22q11 deletion syndrome. Int J Paediatr Dent. 2002;12(1):14–23.
Goldberg R, Motzkin B, Marion R, Scambler PJ, Shprintzen RJ. Velo-cardio-facial syndrome: a review of 120 patients. Am J Med Genet. 1993;45(3):313–9.
Shprintzen RJ, Goldberg RB, Lewin ML, Sidoti EJ, Berkman MD, Argamaso RV, Young D. A new syndrome involving cleft palate, cardiac anomalies, typical facies, and learning disabilities: velo-cardio-facial syndrome. Cleft Palate J. 1978;15(1):56–62.
Zhang M, Li FX, Liu XY, Hou JY, Ni SH, Wang J, Zhao CM, Zhang W, Kong Y, Huang RT, et al. TBX1 loss-of-function mutation contributes to congenital conotruncal defects. Exp Ther Med. 2018;15(1):447–53.
Bartsch O, Nemeckova M, Kocarek E, Wagner A, Puchmajerova A, Poppe M, Ounap K, Goetz P. DiGeorge/velocardiofacial syndrome: FISH studies of chromosomes 22q11 and 10p14, and clinical reports on the proximal 22q11 deletion. Am J Med Genet A. 2003;117A(1):1–5.
Carlson C, Sirotkin H, Pandita R, Goldberg R, McKie J, Wadey R, Patanjali SR, Weissman SM, Anyane-Yeboa K, Warburton D, et al. Molecular definition of 22q11 deletions in 151 velo-cardio-facial syndrome patients. Am J Hum Genet. 1997;61(3):620–9.
Lindsay EA, Goldberg R, Jurecic V, Morrow B, Carlson C, Kucherlapati RS, Shprintzen RJ, Baldini A. Velo-cardio-facial syndrome: frequency and extent of 22q11 deletions. Am J Med Genet. 1995;57(3):514–22.
Kurahashi H, Nakayama T, Osugi Y, Tsuda E, Masuno M, Imaizumi K, Kamiya T, Sano T, Okada S, Nishisho I. Deletion mapping of 22q11 in CATCH22 syndrome: identification of a second critical region. Am J Hum Genet. 1996;58(6):1377–81.
Rump P, de Leeuw N, van Essen AJ, Verschuuren-Bemelmans CC, Veenstra-Knol HE, Swinkels ME, Oostdijk W, Ruivenkamp C, Reardon W, de Munnik S, et al. Central 22q11.2 deletions. Am J Med Genet A. 2014;164A(11):2707–23.
Williams CL, Nelson KR, Grant JH, Mikhail FM, Robin NH. Cleft palate in a patient with the nested 22q11.2 LCR C to D deletion. Am J Med Genet A. 2016;170A(1):260–2.
Baldini A. The 22q11.2 deletion syndrome: a gene dosage perspective. ScientificWorldJournal. 2006;6:1881–7.
Morrow B, Goldberg R, Carlson C, Das Gupta R, Sirotkin H, Collins J, Dunham I, O'Donnell H, Scambler P, Shprintzen R, et al. Molecular definition of the 22q11 deletions in velo-cardio-facial syndrome. Am J Hum Genet. 1995;56(6):1391–403.
Chieffo C, Garvey N, Gong W, Roe B, Zhang G, Silver L, Emanuel BS, Budarf ML. Isolation and characterization of a gene from the DiGeorge chromosomal region homologous to the mouse Tbx1 gene. Genomics. 1997;43(3):267–77.
Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410(6824):97–101.
Zoupa M, Seppala M, Mitsiadis T, Cobourne MT. Tbx1 is expressed at multiple sites of epithelial-mesenchymal interaction during early development of the facial complex. Int J Dev Biol. 2006;50(5):504–10.
Chapman DL, Garvey N, Hancock S, Alexiou M, Agulnik SI, Gibson-Brown JJ, Cebra-Thomas J, Bollag RJ, Silver LM, Papaioannou VE. Expression of the T-box family genes, Tbx1-Tbx5, during early mouse development. Dev Dyn. 1996;206(4):379–90.
Kochilas LK, Potluri V, Gitler A, Balasubramanian K, Chin AJ. Cloning and characterization of zebrafish tbx1. Gene Expr Patterns. 2003;3(5):645–51.
Mitsiadis TA, Tucker AS, De Bari C, Cobourne MT, Rice DP. A regulatory relationship between Tbx1 and FGF signaling during tooth morphogenesis and ameloblast lineage determination. Dev Biol. 2008;320(1):39–48.
Xu H, Morishima M, Wylie JN, Schwartz RJ, Bruneau BG, Lindsay EA, Baldini A. Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development. 2004;131(13):3217–27.
Caton J, Luder HU, Zoupa M, Bradman M, Bluteau G, Tucker AS, Klein O, Mitsiadis TA. Enamel-free teeth: Tbx1 deletion affects amelogenesis in rodent incisors. Dev Biol. 2009;328(2):493–505.
Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27(3):286–91.
Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104(4):619–29.
Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362(9393):1366–73.
Stoller JZ, Epstein JA. Identification of a novel nuclear localization signal in Tbx1 that is deleted in DiGeorge syndrome patients harboring the 1223delC mutation. Hum Mol Genet. 2005;14(7):885–92.
Baldini A. DiGeorge syndrome: the use of model organisms to dissect complex genetics. Hum Mol Genet. 2002;11(20):2363–9.
Scambler PJ. 22q11 deletion syndrome: a role for TBX1 in pharyngeal and cardiovascular development. Pediatr Cardiol. 2010;31(3):378–90.
McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JA, Zackai EH, Emanuel BS, Vermeesch JR, Morrow BE, et al. 22q11.2 deletion syndrome. Nat Rev Dis Primers. 2015;1:15071.
Arnold JS, Braunstein EM, Ohyama T, Groves AK, Adams JC, Brown MC, Morrow BE. Tissue-specific roles of Tbx1 in the development of the outer, middle and inner ear, defective in 22q11DS patients. Hum Mol Genet. 2006;15(10):1629–39.
Xu H, Cerrato F, Baldini A. Timed mutation and cell-fate mapping reveal reiterated roles of Tbx1 during embryogenesis, and a crucial function during segmentation of the pharyngeal system via regulation of endoderm expansion. Development. 2005;132(19):4387–95.
Arvystas M, Shprintzen RJ. Craniofacial morphology in the velo-cardio-facial syndrome. J Craniofac Genet Dev Biol. 1984;4(1):39–45.
Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, Schuffenhauer S, Oechsler H, Belohradsky B, Prieur M, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. 1997;34(10):798–804.
Cohen E, Chow EW, Weksberg R, Bassett AS. Phenotype of adults with the 22q11 deletion syndrome: a review. Am J Med Genet. 1999;86(4):359–65.
Liao J, Kochilas L, Nowotschin S, Arnold JS, Aggarwal VS, Epstein JA, Brown MC, Adams J, Morrow BE. Full spectrum of malformations in velo-cardio-facial syndrome/DiGeorge syndrome mouse models by altering Tbx1 dosage. Hum Mol Genet. 2004;13(15):1577–85.
Gao S, Moreno M, Eliason S, Cao H, Li X, Yu W, Bidlack FB, Margolis HC, Baldini A, Amendt BA. TBX1 protein interactions and microRNA-96-5p regulation controls cell proliferation during craniofacial and dental development: implications for 22q11.2 deletion syndrome. Hum Mol Genet. 2015;24(8):2330–48.
Li C, Lan Y, Jiang R. Molecular and cellular mechanisms of palate development. J Dent Res. 2017;96(11):1184–91.
Gritli-Linde A. The mouse as a developmental model for cleft lip and palate research. Front Oral Biol. 2012;16:32–51.
Lane J, Kaartinen V. Signaling networks in palate development. Wiley Interdiscip Rev Syst Biol Med. 2014;6(3):271–8.
Goudy S, Law A, Sanchez G, Baldwin HS, Brown C. Tbx1 is necessary for palatal elongation and elevation. Mech Dev. 2010;127(5–6):292–300.
Funato N, Nakamura M, Richardson JA, Srivastava D, Yanagisawa H. Tbx1 regulates oral epithelial adhesion and palatal development. Hum Mol Genet. 2012;21(11):2524–37.
Brock LJ, Economou AD, Cobourne MT, Green JB. Mapping cellular processes in the mesenchyme during palatal development in the absence of Tbx1 reveals complex proliferation changes and perturbed cell packing and polarity. J Anat. 2016;228(3):464–73.
Garg V, Yamagishi C, Hu T, Kathiriya IS, Yamagishi H, Srivastava D. Tbx1, a DiGeorge syndrome candidate gene, is regulated by sonic hedgehog during pharyngeal arch development. Dev Biol. 2001;235(1):62–73.
Cobourne MT, Green JB. Hedgehog signalling in development of the secondary palate. Front Oral Biol. 2012;16:52–9.
Parada C, Chai Y. Roles of BMP signaling pathway in lip and palate development. Front Oral Biol. 2012;16:60–70.
Stanier P, Pauws E. Development of the lip and palate: FGF signalling. Front Oral Biol. 2012;16:71–80.
Rice R, Spencer-Dene B, Connor EC, Gritli-Linde A, McMahon AP, Dickson C, Thesleff I, Rice DP. Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J Clin Invest. 2004;113(12):1692–700.
Lan Y, Jiang R. Sonic hedgehog signaling regulates reciprocal epithelial-mesenchymal interactions controlling palatal outgrowth. Development. 2009;136(8):1387–96.
Hu T, Yamagishi H, Maeda J, McAnally J, Yamagishi C, Srivastava D. Tbx1 regulates fibroblast growth factors in the anterior heart field through a reinforcing autoregulatory loop involving forkhead transcription factors. Development. 2004;131(21):5491–502.
Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013;41(Web Server issue):W77–83.
Oliveros JC. VENNY. An interactive tool for comparing lists with Venn Diagrams. 2007. http://bioinfogp.cnb.csic.es/tools/venny/index.html.
Innes PB. The ultrastructure of the mesenchymal element of the palatal shelves of the fetal mouse. J Embryol Exp Morphol. 1978;43:185–94.
Grifone R, Jarry T, Dandonneau M, Grenier J, Duprez D, Kelly RG. Properties of branchiomeric and somite-derived muscle development in Tbx1 mutant embryos. Dev Dyn. 2008;237(10):3071–8.
Kelly RG, Jerome-Majewska LA, Papaioannou VE. The del22q11.2 candidate gene Tbx1 regulates branchiomeric myogenesis. Hum Mol Genet. 2004;13(22):2829–40.
Okano J, Sakai Y, Shiota K. Retinoic acid down-regulates Tbx1 expression and induces abnormal differentiation of tongue muscles in fetal mice. Dev Dyn. 2008;237(10):3059–70.
de Wilde J, Hulshof MF, Boekschoten MV, de Groot P, Smit E, Mariman EC. The embryonic genes Dkk3, Hoxd8, Hoxd9 and Tbx1 identify muscle types in a diet-independent and fiber-type unrelated way. BMC Genomics. 2010;11:176.
Vitelli F, Taddei I, Morishima M, Meyers EN, Lindsay EA, Baldini A. A genetic link between Tbx1 and fibroblast growth factor signaling. Development. 2002;129(19):4605–11.
Sambasivan R, Gayraud-Morel B, Dumas G, Cimper C, Paisant S, Kelly RG, Tajbakhsh S. Distinct regulatory cascades govern extraocular and pharyngeal arch muscle progenitor cell fates. Dev Cell. 2009;16(6):810–21.
Babiarz BS, Allenspach AL, Zimmerman EF. Ultrastructural evidence of contractile systems in mouse palates prior to rotation. Dev Biol. 1975;47(1):32–44.
Wee EL, Zimmerman EF. Palate morphogenesis: II. Contraction of cytoplasmic processes in ATP-induced palate rotation in glycerinated mouse heads. Teratology. 1980;21(1):15–27.
Ivins S, Lammerts van Beuren K, Roberts C, James C, Lindsay E, Baldini A, Ataliotis P, Scambler PJ. Microarray analysis detects differentially expressed genes in the pharyngeal region of mice lacking Tbx1. Dev Biol. 2005;285(2):554–69.
Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1(1):11–21.
Vaziri Sani F, Hallberg K, Harfe BD, McMahon AP, Linde A, Gritli-Linde A. Fate-mapping of the epithelial seam during palatal fusion rules out epithelial-mesenchymal transformation. Dev Biol. 2005;285(2):490–5.
Yoshida M, Shimono Y, Togashi H, Matsuzaki K, Miyoshi J, Mizoguchi A, Komori T, Takai Y. Periderm cells covering palatal shelves have tight junctions and their desquamation reduces the polarity of palatal shelf epithelial cells in palatogenesis. Genes Cells. 2012;17(6):455–72.
Goulet M, Rio M, Jacquette A, Ladouceur M, Bonnet D. Neonatal hypocalcaemic dilated myocardiopathy due to a 22q11 microdeletion. Arch Mal Coeur Vaiss. 2006;99(5):520–2.
Aggarwal VS, Carpenter C, Freyer L, Liao J, Petti M, Morrow BE. Mesodermal Tbx1 is required for patterning the proximal mandible in mice. Dev Biol. 2010;344(2):669–81.
Kim MS, Park MJ, Kim SJ, Lee CH, Yoo H, Shin SH, Song ES, Lee SH. Emodin suppresses hyaluronic acid-induced MMP-9 secretion and invasion of glioma cells. Int J Oncol. 2005;27(3):839–46.
Brown NL, Yarram SJ, Mansell JP, Sandy JR. Matrix metalloproteinases have a role in palatogenesis. J Dent Res. 2002;81(12):826–30.
Zhang Z, Huynh T, Baldini A. Mesodermal expression of Tbx1 is necessary and sufficient for pharyngeal arch and cardiac outflow tract development. Development. 2006;133(18):3587–95.
Economou AD, Ohazama A, Porntaveetus T, Sharpe PT, Kondo S, Basson MA, Gritli-Linde A, Cobourne MT, Green JB. Periodic stripe formation by a Turing mechanism operating at growth zones in the mammalian palate. Nat Genet. 2012;44(3):348–51.
Cobourne MT, Miletich I, Sharpe PT. Restriction of sonic hedgehog signalling during early tooth development. Development. 2004;131(12):2875–85.
Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19(2):185–93.
Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57(1):289–300.
Lan Y, Ovitt CE, Cho ES, Maltby KM, Wang Q, Jiang R. Odd-skipped related 2 (Osr2) encodes a key intrinsic regulator of secondary palate growth and morphogenesis. Development. 2004;131(13):3207–16.
Acknowledgements
The authors are grateful to Antonio Baldini and Peter Scambler for allowing access to the Tbx1 mouse line and Alex Huhn for expert mouse husbandry.
Funding
This work was funded by a European Orthodontic Society Research Grant (to MTC).
Availability of data and materials
The microarray datasets generated and analysed during the current study are available in the Gene Expression Omnibus (GEO) repository at NCBI (GSE37904). https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE37904
Author information
Authors and Affiliations
Contributions
MZ carried out the microarray analysis; in situ hybridization, analysed data and wrote the manuscript; GMX carried out explant culture, in situ hybridization and wrote the manuscript; SB carried out RT-PCR analyses and analysed the data; IT performed bioinformatic analyses and critically revised the manuscript; MA supervised the microarray and validation analyses, analysed data and wrote the manuscript; MTC devised the experiments, analysed the data and wrote the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval
The welfare of animals used in research in the United Kingdom is protected by law. The Animal Scientific Procedures Act 1986 (ASPA) and Amendment Regulations 2012 protects all animals used in procedures for scientific purposes. This act is implemented by the Animals in Science Regulation Unit (ASRU) of the United Kingdom Government Home Office. All animal work was approved by King’s College London Animal Welfare and Ethical Review Body (AWERB) and carried out according to United Kingdom Government Home Office guidelines under project license number PPL70/7866.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Tbx1 lacZ reporter expression in the developing murine palate. (A) E12.5; (B) E13.5; (C) E14.5; (D) E15.5. Tbx1 is expressed in epithelium of the primary (yellow arrowhead) and secondary palate (white arrowhead) with expression persisting in these regions during the process of fusion (orange and pink arrowheads, respectively). Expression is also seen in the maxillary incisor tooth germs (green arrowhead), maxillary molar tooth germs (red arrowhead) and palatal rugae (black arrows). (TIF 2146 kb)
Additional file 2:
List of genes differentially expressed in WT compared to heterozygous palates (n = 400) (Group B). (XLSX 33 kb)
Additional file 3:
KEGG pathway analysis. (XLSX 11 kb)
Additional file 4:
Quantitative RT-PCR primer/probe list. This table contains a complete list of the 63 primers/ probes used in the real-time quantitative RT-PCR analysis of gene expression in the developing palate of Tbx1 mice. (DOCX 90 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zoupa, M., Xavier, G.M., Bryan, S. et al. Gene expression profiling in the developing secondary palate in the absence of Tbx1 function. BMC Genomics 19, 429 (2018). https://doi.org/10.1186/s12864-018-4782-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-4782-y