
Abstract
Background
The therapeutic management of obesity is challenging, hence further elucidating the underlying mechanisms of obesity development and identifying new diagnostic biomarkers and therapeutic targets are urgent and necessary. Here, we performed differential gene expression analysis and weighted gene co-expression network analysis (WGCNA) to identify significant genes and specific modules related to BMI based on gene expression profile data of 7 discordant monozygotic twins.
Results
In the differential gene expression analysis, it appeared that 32 differentially expressed genes (DEGs) were with a trend of up-regulation in twins with higher BMI when compared to their siblings. Categories of positive regulation of nitric-oxide synthase biosynthetic process, positive regulation of NF-kappa B import into nucleus, and peroxidase activity were significantly enriched within GO database and NF-kappa B signaling pathway within KEGG database. DEGs of NAMPT, TLR9, PTGS2, HBD, and PCSK1N might be associated with obesity. In the WGCNA, among the total 20 distinct co-expression modules identified, coral1 module (68 genes) had the strongest positive correlation with BMI (r = 0.56, P = 0.04) and disease status (r = 0.56, P = 0.04). Categories of positive regulation of phospholipase activity, high-density lipoprotein particle clearance, chylomicron remnant clearance, reverse cholesterol transport, intermediate-density lipoprotein particle, chylomicron, low-density lipoprotein particle, very-low-density lipoprotein particle, voltage-gated potassium channel complex, cholesterol transporter activity, and neuropeptide hormone activity were significantly enriched within GO database for this module. And alcoholism and cell adhesion molecules pathways were significantly enriched within KEGG database. Several hub genes, such as GAL, ASB9, NPPB, TBX2, IL17C, APOE, ABCG4, and APOC2 were also identified. The module eigengene of saddlebrown module (212 genes) was also significantly correlated with BMI (r = 0.56, P = 0.04), and hub genes of KCNN1 and AQP10 were differentially expressed.
Conclusion
We identified significant genes and specific modules potentially related to BMI based on the gene expression profile data of monozygotic twins. The findings may help further elucidate the underlying mechanisms of obesity development and provide novel insights to research potential gene biomarkers and signaling pathways for obesity treatment. Further analysis and validation of the findings reported here are important and necessary when more sample size is acquired.
Similar content being viewed by others
Background
Obesity, as a complex disorder mediated by the interplay between genetic and environmental factors [1], has been a public health and policy problem due to its prevalence, costs, and health effects [2]. The therapeutic management of obesity includes lifestyle changes, medications, and surgery. However, the treatment of obesity is challenging because of diverse patient conditions, prolonged and chronic nature of disease, difficulty of maintaining dieting and physical exercise frequently [3,4,5], limited effectiveness and side effects of the medication [6], and high cost and risk of complications of surgery [7]. Other efforts are focused in the development of novel therapeutics, yet the effectiveness requires to be tested and confirmed [8,9,10] and the safety requires to be assessed [11]. Therefore, for the purpose of identifying new diagnostic biomarkers and therapeutic targets and developing novel therapeutic strategies which not only produce sufficient weight loss but also lack side effects, further elucidating the molecular mechanisms underlying obesity development is necessary and urgent.
Recently, gene expression profiling analysis has yielded insights into the measurement of alterations in genetic expression patterns, and has facilitated the identification of differentially expressed genes (DEGs) being crucial to obesity. In a study conducted by Roque DR, et al., obesity related genes, such as LPL, IRS-1, IGFBP4, and IGFBP7, etc., were found to be upregulated with increasing BMI among endometrial cancer patients [12]. The study of Gruchala-Niedoszytko, M, et al. also found a series of genes (PI3, LOC100008589, RPS6KA3, LOC441763, IFIT1, and LOC100133565) with a different expression that may be related to an increased BMI [13]. And genes of PGC1-α, FNCD5, and FGF, which play roles in adipose tissue development and function, were abundantly expressed in subcutaneous, visceral, and epigastric adipose tissues of extreme obesity patients based on gene expression profiling [14]. However, due to the gene expression profiling analysis merely focused on the effect of individual genes and transcripts, without regard to their correlated patterns of expression and the effect of networks of genes, it may fail to detect important biological pathways or gene-gene interactions related to obesity.
Weighted gene co-expression network analysis (WGCNA) is a systems biology method for analyzing the correlation patterns of large and high-dimensional gene expression data sets [15]. It can be used to find modules of highly correlated genes, correlate module eigengenes (MEs) to external sample traits, calculate module membership (MM) and gene significance (GS), and find intramodular hub genes, etc. WGCNA has yielded novel insights into the molecular aspects to identify candidate biomarkers or therapeutic targets. At present, it has been increasingly applied to analyze various gene expression profiles of hepatocellular carcinoma [16], pneumocyte senescence induced by thoracic irradiation [17], psoriasis [18], severe asthma [19], coronary artery disease [20], and lung cancer [21], etc. Although widely being employed, the WGCNA has, to our knowledge, not yet been applied to analyze the expression profiles of BMI-discordant monozygotic twin pairs.
While monozygotic twins are characteristic of the genetic similarity and rearing-environment sharing, they show phenotypic discordance for certain complex traits and diseases. Thus, the discordant monozygotic model is becoming a popular and powerful tool for identifying non-genetic contributions to a phenotype variation including subtle difference in gene expression not mediated by cis- or trans-eQTL effects, and for linking environmental exposure to differential epigenetic regulation while controlling for individual genetic make-up [22,23,24]. Therefore, to reveal the potential molecular mechanisms of obesity, we performed both differential gene expression analysis and WGCNA to analyze the expression profiles of BMI-discordant monozygotic twin pairs. The potentially important DEGs were identified, and the modules correlated with external traits and the hub genes related to BMI were determined. The results may help further elucidate the underlying mechanisms of obesity and provide novel insights to research potential gene biomarkers and signaling pathways for the treatment of obesity.
Methods
Subjects recruitment
The sampling of monozygotic twins was based on the Qingdao Twin Registry at the Qingdao Center for Disease Control and Prevention [25]. Twins were recruited to a clinical investigation after sampling randomly through residence registry and the local disease control network (2012–2013). Written informed consent was obtained from all subjects. We excluded subjects (i) being pregnant or breastfeeding, (ii) undergoing diabetes, (iii) undergoing cardiovascular disease, and (iv) taking any medications within 1 month before participation, and incomplete twin pairs were dropped. The zygosity of twin pairs was determined by DNA testing using 16 short tandem repeat DNA markers. Finally, a total of 7 BMI-discordant monozygotic twin pairs with median age of 52 years (range: 43–65 years) were identified.
For each subject, we took three anthropometric measurements following standard procedures with at least one-minute interval and calculated the mean of these three measurements. Height was measured to the nearest centimeter using a vertical scale with a horizontal moving headboard. And body weight was measured to the nearest 0.1 kg using a standing beam scale. Then BMI was calculated as weight (kg) divided by the square of height (m). Besides, BMI was classified into three classes: Class I, 18.5 ≤ BMI < 24 kg/m2, normal; Class II, 24 ≤ BMI < 28 kg/m2, overweight; and Class III, BMI ≥ 28 kg/m2, obesity. Blood sample was kept frozen at −80 °C for 6 months before sending to routine laboratory testing.
RNA library construction and sequencing and quality control
After total messenger RNA (mRNA) being extracted from whole blood by using TRIzol reagent (Invitrogen, San Diego, USA), the RNA concentration and purity were tested with NanoDrop 2000 Spectrophotometer (Termo Fisher Scientifc, Wilmington, USA) and the RNA integrity was measured with RNA Nano 6000 Assay Kit of Agilent Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, USA).
Then the high-quality RNA was sent to Biomarker Technologies Corporation (Biomarker Technologies Corporation, Beijing, China) for further analysis. The RNA-Seq libraries were constructed with NEBNext UltraTM RNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, USA) following the manufacturer’s recommendations as follows: purifying mRNA with NEBNext Poly (A) mRNA Magnetic Isolation Module, randomly fragmenting isolated mRNA, synthesizing and purifying double-stranded cDNAs, selecting fragment sizes using Agencourt AMPure XP system, and obtaining cDNA library by PCR enrichment. At last, we sequenced the prepared cDNA library using the Illumina HiSeq 2500.
To obtain high-quality clean data (Q30 > 85%), quality control for the raw sequencing data was performed by removing reads containing adapter sequences, unknown nucleotides >5%, and low-quality reads. After mapping to the human genome by TopHat2 [26], we estimated the gene expression levels with fragments per kilobase of exon per million fragments mapped (FPKM) value by Cufflinks software [27].
Differential expression analysis
In the differential expression analysis between the 7 BMI-discordant twin pairs using EBSeq [28], the Benjamini-Hochberg method corrected P-value, i.e., False discovery rate (FDR), was estimated to circumvent false positive results which occurred in the multiple tests [29, 30]. The fold change (FC) of the expression values between twins was also calculated. Then DEGs were defined as those met the criteria of |log2FC| > 1and FDR < 0.01.
Weighted gene co-expression network analysis (WGCNA)
The WGCNA package in R is a comprehensive collection of R functions for performing various aspects of weighted correlation network analysis [15, 31]. Based on the expression profiles of 7 monozygotic twin pairs, the network construction, module detection, module and gene selection, calculations of topological properties, visualization, and interfacing with external software package were conducted following the tutorials provided.
Modules identification
Briefly, after calculating Pearson correlations between each gene pair, we established a weighted adjacency matrix by raising the co-expression similarity to a power β = 29. Subsequently, we constructed the topological overlap matrix (TOM) using correlation expression values [32,33,34]. Then each TOM was used as input for hierarchical clustering analysis [35], and gene modules (i.e. clusters of genes with high topological overlap) was detected by using a dynamic tree cutting algorithm (deep split = 2, cut height = 0.27). The co-expression module structure was visualized by heatmap plots of topological overlap in the gene network. Relationships among modules were summarized by a hierarchical clustering dendrogram of the eigengenes and by a heatmap plot of the corresponding eigengene network.
Relating modules to external traits
To identify modules that were significantly associated with the traits of interest-BMI, BMI classes, and disease status (obesity versus non-obesity), we correlated the MEs (i.e. the first principle component of a module) [36] with external traits and searched the most significant associations.
Hub genes analysis
The MM was defined as the correlation of gene expression profile with ME. And the GS measure was defined as (the absolute value of) the correlation between gene and external traits. Genes with highest MM and highest GS in modules of interest were natural candidates for further research [37,38,39,40]. Thus, the intramodular hub genes were chosen by external traits based GS > 0.2 and MM > 0.8 with a threshold of P-value <0.05 [41]. The gene-gene interaction network was constructed and visualized using VisANT 5.0 [42].
Functional annotation and enrichment analysis
Genes identified in the differential expression analysis and in module of interest in WGCNA were annotated by utilizing BLAST software within the following databases: NCBI nonredundant protein sequences (NR) [43], Clusters of Orthologous Groups (COG) [44], KOG [45], Kyoto Encyclopedia of Genes and Genomes (KEGG) [46, 47], and Gene Ontology (GO) [48, 49]. Subsequently, we drew histogram by mapping GO function of genes in modules of interest to the corresponding secondary features on the background of all genes’ GO annotation. The Pearson Chi-Square test was applied to indicate significant relationships between the two input datasets if all the expected counts were greater than or equal to 5 for 2 × 2 matrixes. And the Fisher’s exact test was applied if one of the expected counts was less than 5. Then we implemented GO enrichment analysis based on a hypergeometric test [50] and calculated a Fisher’s Exact P-Value which was then corrected by Benjamini-Hochberg method. Besides, the KEGG pathways enrichment analysis was conducted by applying the KEGG Orthology-Based Annotation System (KOBAS) utilizing a hypergeometric test [51]. The P-value <0.05 was used as the enrichment cut-off criterion.
Results
Differential expression analysis
A total of 7 BMI-discordant monozygotic twin pairs with median age of 52 years were included for the gene expression profiling analysis (Table 1). The extracted cDNA samples from twins were subjected to sequencing using an Illumina HiSeq2500 platform. The Q30 of each sample was not less than 92.26% and the mapped rate ranged from 87.62% to 93.18% (Additional file 1: Table S1). Under the threshold of |log2FC| > 1 and FDR < 0.01, a range from 360 to 1116 DEGs were identified between co-twin pairs (Table 1). It appeared that 32 DEGs were with a trend of up-regulation in at least three of twins with higher BMI when compared to their siblings (Additional file 2: Table S2). Of these, three genes were found up-regulated in 4 twin pairs, and the others were found up-regulated in 3 twin pairs.
As the summarized results of enrichment analysis within GO and KEGG databases shown (Table 2), several potentially important findings emerged (Corrected P-value < 0.05), including positive regulation of nitric-oxide synthase biosynthetic process (P = 5.34E-03), positive regulation of NF-kappa B import into nucleus (P = 1.04E-02), peroxidase activity (P = 6.82E-03), and NF-kappa B signaling pathway (P = 4.49E-02). Genes of NAMPT, TLR9, PTGS2, HBD, and BCL2L1 were involved in these significant findings. In addition, PCSK1N gene might also be associated with obesity. We compared previously implicated BMI-related gene expression differences in study of Homuth, G, et al. [52] with ours to validate the findings further. This comparison revealed consistency for positive BMI-associated expression differences, including HBD, XK, SELENBP1, SNCA, LAS2, PLEK2, GLRX5, TMOD1, SLC4A1, BCL2L1, TRIM58, DCAF12, NFIX, BSG, PLVAP, and PCSK1N.
WGCNA
Modules identification
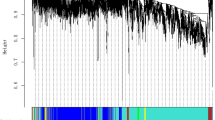
WGCNA was applied to investigate gene sets that were related to traits of interest-BMI, BMI classes, and disease status using the gene expression data of 7 monozygotic twin pairs. After using a dynamic tree cutting algorithm, a total of 20 distinct co-expression modules containing 48 to 9274 genes per module were identified, and 1912 uncorrelated genes were assigned into a grey module which was ignored in the following study (Fig. 1, and Additional file 3: Table S3). The heatmap plot of topological overlap in the gene network is depicted (Fig. 2).

Gene dendrogram obtained by average linkage hierarchical clustering. The color row underneath the dendrogram shows the assigned original module and the merged module

Heatmap plot of topological overlap in the gene network. In the heatmap, each row and column corresponds to a gene, light color denotes low topological overlap, and progressively darker red denotes higher topological overlap. Darker squares along the diagonal correspond to modules. The gene dendrogram and module assignment are shown along the left and top
Relating modules to external traits
To understand the physiologic significance of the modules, we correlated the 20 MEs with traits of interest and searched for the most significant associations. According to the heatmap of module-trait correlation (Fig. 3), genes clustered in coral1 module (68 genes) had the strongest positive correlation with BMI (r = 0.56, P = 0.04) and disease status (r = 0.56, P = 0.04), whereas statistically nonsignificant correlation was found with BMI classes (r = 0.51, P = 0.06). Nevertheless, the ME of saddlebrown module (212 genes) was only significantly correlated with BMI (r = 0.56, P = 0.04). Thus, we would mainly consider coral1 module in the following because this module may indicate external traits more accurately. None of the other modules had a significant association with external traits.

Relationships of consensus module eigengenes and external traits. Each row in the table corresponds to a consensus module, and each column to a sample or trait. Numbers in the table report the correlations of the corresponding module eigengenes and traits, with the P-values printed below the correlations in parentheses. The table is color coded by correlation according to the color legend
Relationships among modules
To study the relationships among modules and determine their correlation with trait of BMI, we correlated the MEs. The eigengene network using a dendrogram and a heatmap plot are depicted in Fig. 4. The dendrogram (Fig. 4a) indicated that the coral1 and saddlebrown modules were highly correlated, and trait of BMI fell within the meta-module grouping together the two modules. The heatmap plot (Fig. 4b) showed the detailed eigengenes adjacencies of all modules and trait of BMI.

Relationships among modules. a Hierarchical clustering of module eigengenes that summarize the modules found in the clustering analysis. Branches of the dendrogram (the meta-modules) group together eigengenes that are positively correlated. b Heatmap plot of the adjacencies in the eigengene network including the trait of interest-BMI. Each row and column in the heatmap corresponds to one module eigengene (labeled by color) or BMI. In the heatmap, red represents high adjacency, while blue color represents low adjacency. Squares of red color along the diagonal are the meta-modules
Functional annotation and enrichment analysis for coral1 module
In order to provide an interpretation of the biological mechanism associated with the genes clustered in module of interest--coral1, we conducted functional annotation (Additional file 4: Table S4) and enrichment analysis.
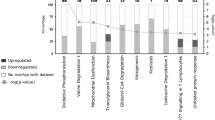
Three main annotated categories-biological process, cellular component, and molecular function were obtained in GO database (Fig. 5, and Additional file 5: Table S5). The proportion of genes in coral1 module increased significantly in certain subgroups, including single-organism process (P = 0.024), multicellar organismal process (P = 0.004), developmental process (P = 0.009), localization (P = 0.002), signaling (P = 0.005), extracellar region (P = 0.009), and transporter activity (P = 0.023).

GO classification in coral1 module. Annotation statistics of genes in the secondary node of GO. The horizontal axis shows secondary nodes of three categories in GO. The vertical axis displays the percentage of annotated genes versus the total gene number. The left columns display annotation information of the total genes and the right columns represent annotation information of the genes clustered in coral1 module
As the summarized results of enrichment analysis within GO database shown (Table 3), several potentially important findings emerged (Corrected P-value < 0.05). In the biological processes, categories of positive regulation of phospholipase activity (P = 2.91E-03), high-density lipoprotein particle clearance (P = 4.36E-03), chylomicron remnant clearance (P = 4.36E-03), reverse cholesterol transport (P = 2.24E-02), and positive regulation of axon extension (P = 2.61E-02) were significantly enriched. Among the 6 enrichment categories in the cellular component, intermediate-density lipoprotein particle (P = 2.74E-03), chylomicron (P = 9.97E-03), low-density lipoprotein particle (P = 1.89E-02), and very-low-density lipoprotein particle (P = 2.74E-02) were related to lipid transport and metabolism. And categorie of voltage-gated potassium channel complex (P = 1.43E-02) may be potentially involved in the regulating energy homeostasis. In the molecular function, the categories of cholesterol transporter activity (P = 9.72E-03) and neuropeptide hormone activity (P = 1.98E-02) should also be highlighted.
As shown in Additional file 6: Table S6, the KEGG annotation results were classified according to KEGG pathway classification. Two pathways of alcoholism and cell adhesion molecules (CAMs) were significantly enriched in KEGG database (Table 3). And the COG function classification results are shown in Additional file 7: Table S7.
Hub genes analysis in coral1 module
Figure 6 shows the scatterplots of GS for traits of BMI, BMI classes, and disease status versus MM in coral1 module. MM and GS for BMI (Fig. 6a), BMI classes (Fig. 6b), and disease status (Fig. 6c) exhibited very significant positive correlations, implying that the most important (central) elements of coral1 module also tended to be highly correlated with these external traits. The identified 21 hub genes (Additional file 8: Table S8) included GAL, ASB9, KCNT1, NPPB, TBX2, KCNK15, IL17C, APOE, LBX1, LRRC38, LINGO1, ABCG4, LCN15, RFLNA, SOX18, C1orf146, APOC2, PRSS29P, LOC102724223, C7orf71, and IGKV1D-17. The visualized plot of the gene-gene interaction network in coral1 module is shown in Fig. 7.

Scatterplots of gene significance (GS) for external traits versus module membership (MM) in the coral1 module. MM and GS for BMI, BMI classes, and disease status exhibit very significant correlations, implying that the most important (central) elements of coral1 module also tend to be highly correlated with these external traits. a Module membership vs. gene significance for BMI; (b) Module membership vs. gene significance for BMI classes; and (c). Module membership vs. gene significance for disease status

Interaction of gene co-expression patterns by VisANT 5.0 in the coral1 module. The node size and edge number are proportional to degree and connection strength, respectively. Eight red nodes indicate the hub genes potentially related to BMI in the coral1 module. Among the 8 genes, GAL, APOE, APOC2, and NPPB have been demonstrated to be associated with obesity and the others would be associated with obesity as the related works suggested
The 21 hub genes were involved in several enriched functional items (Table 3), including high-density lipoprotein particle clearance, chylomicron remnant clearance, phospholipid efflux, reverse cholesterol transport, chylomicron, voltage-gated potassium channel complex, very-low-density lipoprotein particle, and cholesterol transporter activity, most of which were associated with lipid transport and metabolism. None of the 68 genes in coral1 module was identified as DEGs.
Saddlebrown module
In the functional annotation analysis within GO database, the proportion of genes in saddlebrown module increased in subgroups of extracellular region (P = 0.002) and extracellular region part (P = 0.019) (Additional file 9: Figure S1, and Additional file 10: Table S9). The categories of structural constituent of eye lens (P = 1.14E-02) and troponin T binding (P = 2.75E-02) were significantly enriched in the molecular function, whereas no categories were significantly enriched in biological process and cellular component. The results of KEGG pathway classification and COG function classification are shown in Additional file 11: Table S10 and Additional file 12: Table S11, respectively. BMI based GS and MM exhibited a very significant correlation in saddlebrown module (Additional file 13: Figure S2), and hub genes of KCNN1, CNN1, and AQP10 were identified as DEGs.
Discussion
In the differential expression analysis based on the gene expression data of 7 BMI-discordant monozygotic twin pairs, we identified 32 genes with a trend of up-regulation in twins with higher BMI when compared to their siblings. Several potentially important enrichment findings emerged, including positive regulation of nitric-oxide synthase biosynthetic process, positive regulation of NF-kappa B import into nucleus, peroxidase activity, and NF-kappa B signaling pathway (Table 2). And up-regulated genes-NAMPT, TLR9, PTGS2, HBD, and PCSK1N might be associated with obesity risk. In addition, we also applied WGCNA to quantitatively analyze the interconnectedness of gene expression data and assessed the importance of genes within the networks. Among the 20 distinct co-expression modules identified, genes clustered in coral1 module had the strongest positive correlation with BMI and disease status (Fig. 3), indicating that the highly co-expressed genes in this module had potential biological significance. Functional enrichment analysis revealed several significant enrichments of BMI-related categories for coral1 module. Importantly, several hub genes were strongly related to lipid transport and metabolism (Table 3) and may be particularly valuable for identifying the candidate biomarkers and therapeutic targets for obesity assessed by BMI. Besides, the ME of saddlebrown module was also significantly correlated with BMI (Fig. 3) and 3 hub genes were identified as DEGs.
Obesity is a complex disease under the control of both genetic and environmental factors through the interface of epigenetics, where different combinations of genetic and epigenetic variations can lead to a common phenotype. Considering this, we would not necessarily expect each of the 7 BMI-discordant monozygotic twin pairs to present exactly the same series of gene aberrations, and the stringency of the criterion on the commonality of gene changes was relaxed. In the differential expression analysis, it appeared that 32 genes were with a trend of up-regulation in at least three of twins with higher BMI when compared to their siblings (Additional file 2: Table S2). Besides, four potentially important enrichment findings emerged and 5 up-regulated genes associated with obesity risk were identified as follows (Table 2).
Nitric oxide (NO), whose production is mostly through the action of the nitric oxide synthase (NOS) family of enzymes, is emerging as a central regulator of energy metabolism and body composition. The isoform of inducible nitric oxide synthase (iNOS)-derived NO can promote insulin resistance and inflammation in key peripheral tissues such as liver, skeletal muscle, and adipose tissue. In addition, iNOS may affect glucose homeostasis. Thus, the iNOS isoform appears to promote deleterious changes in metabolism [53]. Considering this, the two up-regulated genes (NAMPT and TRL9) involved in the category of positive regulation of nitric-oxide synthase biosynthetic process should be considered notably (Table 2). And it is indicated that the protein of NAMPT gene can directly activate pathways leading to iNOS induction [54].
It has been revealed that a characteristic feature of obesity linking it to insulin resistance is the presence of chronic low-grade inflammation which is indicative of activation of the innate immune system. The IKK/NF-κB pathway is a well-known inflammatory signaling pathway involved in the pathogenesis of obesity [55, 56], and the two genes-PTGS2 and TLR9 involved in this enrichment term should also be focused (Table 2). In addition, the protein encoded by PTGS2 gene is indicated to be linked with energy homeostasis and metabolic processes based on a cohort of children presenting with syndromic obesity [57]. Even though both these two genes were enriched to the NF-kappa B signaling pathway in KEGG database (Table 2), PTGS2 gene was involved in the inflammation process while BCL2L1 gene might be related to survival process.
In mammals, the peroxidases comprise 8 glutathione peroxidases (GPx1–GPx8) so far identified. Too much data regarding the association between obesity and GPx1, GPx3, GPx4, and GPx7 has been reported [58]. Thus, the two genes of PTGS2 and HBD could be regard as the candidates for further research (Table 2).
Moreover, SNPs in or near PCSK1 loci may also contribute to obesity risk [59, 60]. The associations with BMI for other DEGs should be explored further.
To validate the identified DEGs further, we compared previously implicated BMI-related gene expression differences [52] with ours. This comparison revealed consistency for positive BMI-associated expression differences including HBD, XK, SELENBP1, SNCA, LAS2, PLEK2, GLRX5, TMOD1, SLC4A1, BCL2L1, TRIM58, DCAF12, NFIX, BSG, PLVAP, and PCSK1N. Two consistent genes (SNCA and DCAF12) were also revealed when compared with the BMI-related genes by the Data-driven Expression Prioritized Integration for Complex Traits (DEPICT) method.
In the WGCNA, the proportion of genes in coral1 module increased significantly in subgroups of developmental process, signaling, extracellar region, and transporter activity (Fig. 5, and Additional file 5: Table S5), indicating that these functions may be associated with metabolism and accelerated growth and development of obesity individuals. Notably, GO enrichment analysis also provided more significant results with more biological meanings as follows (Table 3).
Obese subjects frequently suffer atherogenic dyslipidemia which is commonly manifested as elevated plasma free fatty acids, triglycerides (TG) and very low-density lipoprotein (VLDL) levels, decreased high-density lipoprotein cholesterol (HDL-C) levels, and abnormal low-density lipoprotein cholesterol (LDL-C) [61, 62]. Our study suggested that categories involved in lipid metabolism and transport were significantly enriched within the coral1 module, including high-density lipoprotein particle clearance, reverse cholesterol transport, low-density lipoprotein particle, cholesterol transporter activity, chylomicron, chylomicron remnant clearance, very-low-density lipoprotein particle, and intermediate-density lipoprotein particle (Table 3).
Category of positive regulation of phospholipase activity was also enriched in the coral1 module (Table 3). Phospholipids were identified as potential biomarkers for obesity [63]. And it was reported that members of the phospholipase A2 (PLA2) family of enzymes, such as PLA2G1B [64], PLA2G5, and PLA2G2E [65], can serve a distinct role in generating active lipid metabolites, which can promote inflammatory metabolic diseases including obesity [66, 67]. In addition, AdPLA enzyme in white adipose tissue can function as a regulator of lipolysis through increasing prostaglandin E2 (PGE2) formation and decreasing intracellular cAMP [68].
The hypothalamic peptides, such as neuropeptide Y (NPY) and melanin concentrating hormone (MCH) [69], and the peripheral neuropeptides, such as hormone leptin [70], play important roles in regulating food intake and maintaining energy balance [71]. Normally, a dynamic equilibrium exists between orexigenic peptides and anorexigenic peptides [70]. And after receiving stimulus information of neural signal, hormone signal, and metabolites, etc., the hypothalamus appestat maintains the dynamic equilibrium of energy by neuro-humoral response. Therefore, the enriched category of neuropeptide hormone activity may also exert a significant meaning regarding obesity (Table 3).
Another significant GO enriched category was voltage-gated potassium channel (Kv) complex (Table 3). Studies had suggested the relation of subtype-Kv1.3 to insulin sensitivity and the participation of Kv1.3 in regulating energy homeostasis and body weight [72, 73]. Hence, Kv1.3 may be a putative and promising pharmacological target for the treatment of obesity, type II diabetes mellitus and related metabolic diseases [72].
Two pathways of alcoholism and cell adhesion molecules (CAMs) were significantly enriched in KEGG database (Table 3). A growing body of literatures indicate that overlapping central pathways may be involved with uncontrolled eating and excessive ethanol drinking [74]. And emerging link between familial alcoholism risk and obesity in women and possibly in men is identified in recent years [75]. Furthermore, some genetic variants are associated with both alcohol dependence and obesity [76, 77]. Therefore, the genes involved in the alcoholism pathway may be used as potential links between alcoholism and obesity, and as promising targets for controlling ethanol abuse and food intake. As for the CAMs, a review concluded that anthropometric indicators, body composition and eating pattern positively modulate the subclinical inflammation of obesity through reducing CAMs and chemokines [78]. Moreover, a recent study also identified the relationship of adiposity to several CAMs [79].
We visualized the gene-gene interaction network in coral1 module to obtain an insight on the hidden mechanisms (Fig. 7), and a total of 21 hub genes were identified (Additional file 8: Table S8). The hub genes were involved in various gene families and might serve as candidates for additional mechanistic studies and therapeutic interventions. Hub genes of GAL, APOE, APOC2, and NPPB have been demonstrated to be associated with obesity as follows: (I) GAL: Galanin peptides, as the protein for GAL, is undoubtedly involved in the regulation of food intake and body weight. It has been identified that both central galanin and peripheral galanin can affect appetite, food intake and body weight of animals, and the latter can also affect gastrointestinal motility and brown adipose tissue activity [80, 81]. Particularly, newly discovered galanin-like peptide (GALP) may play a role in boosting appetite, body weight, and obesity [80, 82]. Overall, galanin and its receptors may serve as a novel anti-obesity strategy in the future. (II) APOE: ApoE synthesized by adipocyte is a polymorphic glycoprotein in humans, and is a major constituent of HDL, VLDL, and remnant lipoproteins (RLPs). ApoE was identified playing an important role in the development of obesity and insulin resistance in experimental mouse models [83], and the mutation in APOE was involved in lipid metabolism [84] and lipid levels [85] in population studies. Moreover, an equally vital role in adipocyte triglyceride accumulation and VLDL-induced adipogenesis was summarized [86]. Overall, it has been identified that APOE expression serves as a key peripheral contributor to the development of obesity and related metabolic dysfunctions [83, 87]. (III) APOC2: Apolipoprotein C-II (ApoC-II), as a constituent of chylomicrons, VLDL, LDL and HDL, is a cofactor for lipoprotein lipase, which can hydrolyze TG. The gene APOC2 mutation can result in hypertriglyceridemia, which is one of the main characteristics of obese subjects [88]. Besides, an excess of ApoC-II is related to increase of triglyceride-rich particles and alterations in HDL particle distribution [89]. (IV) NPPB: A growing body of evidence indicates that the natriuretic peptides (NPs) system holds the potential to be amenable to therapeutical intervention against obesity. Vila, G, et al. demonstrate that B-Type Natriuretic Peptide (BNP) plays an important role in reducing circulating ghrelin concentrations, decreasing hunger, and increasing feeling of satiety in healthy individuals [90]. Moreover, the function of enhancing lipolysis and energy expenditure, and modulating adipokine release and food intake is also identified [91]. In addition, one recent review emphasized the ability of NPs to regulate body weight and energy homeostasis by driving lipolysis, facilitating beiging of adipose tissues, and promoting lipid oxidation and mitochondrial respiration [92]. Moreover, another review drew the similar conclusion [93].
Although there was no strong indication that TBX2, ASB9, IL17C, and ABCG4 were the causal variant of obesity in the population, studies showed that these genes may also be part of the multifactorial etiology of this complex condition as follows: (I) TBX2: The results of a prospective cohort on the associations of menarche-related genetic variants with pubertal growth in adolescents indicated that SNPs (rs757608) near TBX2 is associated with a rapid weight gain [94]. (II) ASB9: It indicated that overexpression of ASB9 can induce ubiquitination of ubiquitous mitochondrial creatine kinase (uMtCK) [95] in a specific, SOCS box-independent manner [96]. The intracellular creatine kinase (CK) system may be involved in the storage of fat and the development of obesity [97,98,99]. Besides, one cross-sectional study recently provided further evidence that CK may play a role in the pathophysiology of obesity and serve as a marker to identify individuals at risk for obesity [100]. (III) IL17C: Obesity, in some sense, is considered to be an inflammatory predisposition. And interleukin-17 (IL-17) may impact adipose tissue due to the association with induction of tissue inflammation. Particularly, the potential implications of IL-17 in relation to obesity has been consolidated by Ahmed, M and Gaffen, SL [101]. And one study also suggested a linear negative association between IL-17 and visceral adipose tissue thickness [102]. However, the exact role of IL-17C in obesity remains to be explored. (IV) ABCG4: An additional hub gene that should be further investigated is ABCG4, one member of the ABCG family. Studies indicated that ABCG4 promotes cholesterol efflux from cells to HDL [103, 104].
Even though no sufficient studies showed the association of two genes of KCNT1 and LBX1 with obesity, the results of functional annotation and enrichment analysis indicated that they were involved in intermediate-density lipoprotein particle and voltage-gated potassium channel complex, respectively. Thus, they may also be regarded as the targets for etiology research of obesity. Other hub genes in coral1 module were of unknown function in terms of obesity currently, whereas they may also be interesting potential candidates to be future researched and validated.
Among the hub genes in saddlebrown module, KCNN1, CNN1, and AQP10 were up-regulated with increasing BMI in twins. (I) KCNN1: The lipotoxicity in morbid obesity can gradually impair insulin action in the liver and muscle, aggravating insulin resistance [105], and Ye, J proposed an energy-based concept of insulin resistance, in which insulin resistance is a result of energy surplus in cells [106]. The protein of gene KCNN1--small conductance calcium-activated potassium channel protein 1, can serve as a key regulator of excitability and endocrine function in beta cells [107]. (II) AQP10: Aquaglyceroporins, such as AQP10, represent novel additional pathways for the transport of glycerol in human adipocytes [108], and the deregulation in the expression of aquaglyceroporins in adipose tissue is associated with human obesity [109, 110].
Several strengths must be noticed in our study. First, gene expression levels may be under the effect of subjects’ genetic background, gender, age, and environmental exposures as well as by some experimental variables related to clinical sampling, processing, and data analysis. However, the discordant monozygotic model, which is characteristic of the genetic similarity and rearing-environment sharing, is becoming a popular and powerful tool for identifying non-genetic contributions to a phenotype variation including difference in gene expression. Hence, our results of WGCNA, based on the expression data generated from BMI-discordant monozygotic model, may be more credible. Another strength of our study was that the WGCNA provides information on the correlated patterns of expression and the effect of networks of genes, which is useful for detecting important biological pathways or gene-gene interactions related to obesity. Specifically, a set of genes sharing similar functions and correlated to one another in coral1 and saddlebrown modules were identified in our study, some of which have already been verified to play efficient roles in obesity.
Nevertheless, our study has potential limitations as well. First, our study was with small sample size and limited statistical power resulting from the challenges of identifying and recruiting qualified monozygotic twins discordant for BMI. The BMI-discordant monozygotic model, however, helps to mitigate confounding factors associated with genetic polymorphisms in studies of unrelated human subjects and to identify non-genetic contributions to a phenotype variation including difference in gene expression. Besides, we had identified significant genes and specific modules potentially related to BMI. It’s still important and necessary to validate our findings when more sample size is acquired. Second, we couldn’t validate our results with an external and independent dataset because of lacking public BMI-discordant monozygotic dataset with adequate size currently. However, we compared previously implicated BMI-related gene expression differences with our findings, and 16 consistent positive BMI-associated findings were revealed. Third, some genes may be involved in multiple processes/functions which require different gene sets. However, it was difficult to characterize such gene interactions because of the impossibility of forming overlapping modules by WGCNA. Fourth, as in any other studies based on microarray technology, changes in protein levels may not reflect similar changes in mRNA levels accurately because post-translational modification also acts importantly in controlling biological processes. Hence, it may be necessary to validate our results by other techniques. And fifth, in saddlebrown module, the 3 hub genes also identified as DEGs were differentially expressed in just one twin pair. More studies are needed to confirm these results.
Conclusions
In summary, we identified 32 DEGs with a trend of up-regulation in twins with higher BMI when compared to their siblings in the differential expression analysis and determined one module most positively correlated with BMI and several hub genes in the WGCNA. The potentially significant genes and pathways correlated with BMI identified in our analysis may help further elucidate the molecular mechanisms underlying obesity development and provide novel insights regarding future prognostic and therapeutic approaches. Further analysis and validation of the candidate biomarkers of obesity reported here are necessary, including those that have not yet been definitely identified.
Abbreviations
- BMI:
-
Body mass index
- DEGs:
-
Differentially expressed genes
- FC:
-
Fold change
- FDR:
-
False discovery rate
- GO:
-
Gene Ontology
- GS:
-
Gene significance
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- MM:
-
Module membership
- WGCNA:
-
Weight gene co-expression network analysis
References
Paquot N, De Flines J, Rorive M. Obesity: a model of complex interactions between genetics and environment. Revue medicale de Liege. 2012;67:332–6.
U.S. Department of Health and Human Services. The surgeon General’s call to action to prevent and decrease overweight and obesity. Rockville: U.S. Department of Health and Human Services, Public Health Service, Office of the Surgeon General; 2001.
Strychar I. Diet in the management of weight loss. CMAJ. 2006;174:56–63.
Shick SM, Wing RR, Klem ML, McGuire MT, Hill JO, Seagle H. Persons successful at long-term weight loss and maintenance continue to consume a low-energy, low-fat diet. J Am Diet Assoc. 1998;98:408–13.
Tate DF, Jeffery RW, Sherwood NE, Wing RR. Long-term weight losses associated with prescription of higher physical activity goals. Are higher levels of physical activity protective against weight regain? Am J Clin Nutr. 2007;85:954–9.
Rucker D, Padwal R, Li SK, Curioni C, Lau DC. Long term pharmacotherapy for obesity and overweight: updated meta-analysis. BMJ. 2007;335:1194–9.
Colquitt JL, Pickett K, Loveman E, Frampton GK. Surgery for weight loss in adults. Cochrane Database Syst Rev. 2014. Art. No.: CD003641. doi:10.1002/14651858.CD003641.pub4.
Jayasinghe TN, Chiavaroli V, Holland DJ, Cutfield WS, O'Sullivan JM. The new era of treatment for obesity and metabolic disorders: evidence and expectations for gut microbiome transplantation. Front Cell Infect Microbiol. 2016;6:15.
Fanelli RD, Andrew BD. Is Endoluminal bariatric therapy a new paradigm of treatment for obesity? Clin Gastroenterol H. 2016;14:507–15.
Sukhdev S, Bhupender S, Singh KS. Pharmacotherapy & Surgical Interventions Available for obesity management and importance of pancreatic lipase inhibitory Phytomolecules as safer anti-obesity therapeutics. Mini Rev Med Chem. 2017;17:371–9.
Halpern B, Mancini MC. Safety assessment of combination therapies in the treatment of obesity: focus on naltrexone/bupropion extended release and phentermine-topiramate extended release. Expert Opin Drug Saf. 2017;16:27–39.
Roque DR, Makowski L, Chen TH, Rashid N, Hayes DN, Bae-Jump V. Association between differential gene expression and body mass index among endometrial cancers from the cancer genome atlas project. Gynecol Oncol. 2016;142:317–22.
Gruchala-Niedoszytko M, Niedoszytko M, Sanjabi B, van der Vlies P, Niedoszytko P, Jassem E, et al. Analysis of the differences in whole-genome expression related to asthma and obesity. Pol Arch Med Wewn. 2015;125:722–30.
Gerhard GS, Styer AM, Strodel WE, Roesch SL, Yavorek A, Carey DJ, et al. Gene expression profiling in subcutaneous, visceral and epigastric adipose tissues of patients with extreme obesity. Int J Obesity. 2014;38:371–8.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9:559.
Zhang J, Baddoo M, Han C, Strong MJ, Cvitanovic J, Moroz K, et al. Gene network analysis reveals a novel 22-gene signature of carbon metabolism in hepatocellular carcinoma. Oncotarget. 2016;7:49232–45.
Xing Y, Zhang J, Lu L, Li D, Wang Y, Huang S, et al. Identification of hub genes of pneumocyte senescence induced by thoracic irradiation using weighted gene coexpression network analysis. Mol Med Rep. 2016;13:107–16.
Sundarrajan S, Arumugam M. Weighted gene co-expression based biomarker discovery for psoriasis detection. Gene. 2016;593:225–34.
Modena BD, Bleecker ER, Busse WW, Erzurum SC, Gaston BM, Jarjour NN, et al. Gene Expression Correlated to Severe Asthma Characteristics Reveals Heterogeneous Mechanisms of Severe Disease. Am J Respir Crit Care Med. 2016;195:1449-63.
Liu J, Jing L, Tu X. Weighted gene co-expression network analysis identifies specific modules and hub genes related to coronary artery disease. BMC Cardiovasc Disord. 2016;16:54.
Guo Y, Xing Y. Weighted gene co-expression network analysis of pneumocytes under exposure to a carcinogenic dose of chloroprene. Life Sci. 2016;151:339–47.
Castillo-Fernandez JE, Spector TD, Bell JT. Epigenetics of discordant monozygotic twins: implications for disease. Genome Med. 2014;6:60.
Tan Q, Christiansen L, von Bornemann Hjelmborg J, Christensen K. Twin methodology in epigenetic studies. J Exp Biol. 2015;218:134–9.
Zhang D, Li S, Tan Q, Pang Z. Twin-based DNA methylation analysis takes the center stage of studies of human complex diseases. J Genet Genomics. 2012;39:581–6.
Pang Z, Ning F, Unger J, Johnson CA, Wang S, Guo Q, et al. The Qingdao twin registry: a focus on chronic disease research. Twin Res Hum Genet. 2006;9:758–62.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5.
Leng N, Dawson JA, Thomson JA, Ruotti V, Rissman AI, Smits BM, et al. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics. 2013;29:1035–43.
Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J Roy Stat Soc B. 1995;57:289–300.
Benjamini Y. Discovering the false discovery rate. J R Stat Soc B. 2010;72:405–16.
Langfelder P, Horvath S. Fast R functions for robust correlations and hierarchical clustering. J Stat Softw. 2012;46
Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17.
Yip AM, Horvath S. Gene network interconnectedness and the generalized topological overlap measure. BMC Bioinform. 2007;8:22.
Li A, Horvath S. Network neighborhood analysis with the multi-node topological overlap measure. Bioinformatics. 2007;23:222–31.
Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabasi AL. Hierarchical organization of modularity in metabolic networks. Science. 2002;297:1551–5.
Langfelder P, Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst Biol. 2007;1:54.
Horvath S, Zhang B, Carlson M, Lu KV, Zhu S, Felciano RM, et al. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc Natl Acad Sci U S A. 2006;103:17402–7.
Ghazalpour A, Doss S, Zhang B, Wang S, Plaisier C, Castellanos R, et al. Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet. 2006;e130:2.
Fuller TF, Ghazalpour A, Aten JE, Drake TA, Lusis AJ, Horvath S. Weighted gene coexpression network analysis strategies applied to mouse weight. Mamm Genome. 2007;18:463–72.
Oldham MC, Horvath S, Geschwind DH. Conservation and evolution of gene coexpression networks in human and chimpanzee brains. Proc Natl Acad Sci U S A. 2006;103:17973–8.
Horvath S, Dong J. Geometric interpretation of gene coexpression network analysis. PLoS Comput Biol. 2008;4:e1000117.
VisANT 5.0. Visual analyses of metabolic networks in cells and ecosystems. http://visant.bu.edu. Accessed 19 Apr 2017.
Deng YY, Li JQ, Wu SF, Zhu YP, Chen YW, He FC. Integrated nr database in protein annotation system and its localization. Comput Eng. 2006;32:71–4.
Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–6.
Koonin EV, Fedorova ND, Jackson JD, Jacobs AR, Krylov DM, Makarova KS, et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004;5:R7.
Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–80.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30.
Tweedie S, Ashburner M, Falls K, Leyland P, McQuilton P, Marygold S, et al. FlyBase: enhancing drosophila gene ontology annotations. Nucleic Acids Res. 2009;37:D555–9.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000;25:25–9.
Falcon S. Gentleman R. Hypergeometric testing used for gene set enrichment analysis. Bioconductor case studies. New York: Springer New York; 2008. p. 207–20.
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39:W316–22.
Homuth G, Wahl S, Muller C, Schurmann C, Mader U, Blankenberg S, et al. Extensive alterations of the whole-blood transcriptome are associated with body mass index: results of an mRNA profiling study involving two large population-based cohorts. BMC Med Genet. 2015;8:65.
Sansbury BE, Hill BG. Regulation of obesity and insulin resistance by nitric oxide. Free Radic Biol Med. 2014;73:383–99.
Romacho T, Azcutia V, Vazquez-Bella M, Matesanz N, Cercas E, Nevado J, et al. Extracellular PBEF/NAMPT/visfatin activates pro-inflammatory signalling in human vascular smooth muscle cells through nicotinamide phosphoribosyltransferase activity. Diabetologia. 2009;52:2455–63.
Kalin S, Heppner FL, Bechmann I, Prinz M, Tschop MH, Yi CX. Hypothalamic innate immune reaction in obesity. Nat Rev Endocrinol. 2015;11:339–51.
Ringseis R, Eder K, Mooren FC, Kruger K. Metabolic signals and innate immune activation in obesity and exercise. Exerc Immunol Rev. 2015;21:58–68.
Vuillaume ML, Naudion S, Banneau G, Diene G, Cartault A, Cailley D, et al. New candidate loci identified by array-CGH in a cohort of 100 children presenting with syndromic obesity. Am J Med Genet A. 2014;164a:1965–75.
Picklo MJ, Long EK, Vomhof-DeKrey EE. Glutathionyl systems and metabolic dysfunction in obesity. Nutr Rev. 2015;73:858–68.
Hsiao TJ, Hwang Y, Chang HM, Lin E. Association of the rs6235 variant in the proprotein convertase subtilisin/kexin type 1 (PCSK1) gene with obesity and related traits in a Taiwanese population. Gene. 2014;533:32–7.
Rouskas K, Kouvatsi A, Paletas K, Papazoglou D, Tsapas A, Lobbens S, et al. Common variants in FTO, MC4R, TMEM18, PRL, AIF1, and PCSK1 show evidence of association with adult obesity in the Greek population. Obesity (Silver Spring). 2012;20:389–95.
Schmidt AM. The growing problem of obesity: mechanisms, consequences, and therapeutic approaches. Arterioscler Thromb Vasc Biol. 2015;35:e19–23.
Jung UJ, Choi MS. Obesity and its metabolic complications: the role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int J Mol Sci. 2014;15:6184–223.
Rauschert S, Uhl O, Koletzko B, Hellmuth C. Metabolomic biomarkers for obesity in humans: a short review. Ann Nutr Metab. 2014;64:314–24.
Cash JG, Kuhel DG, Goodin C, Hui DY. Pancreatic acinar cell-specific overexpression of group 1B phospholipase A2 exacerbates diet-induced obesity and insulin resistance in mice. Int J Obes. 2011;35:877–81.
Sato H, Taketomi Y, Ushida A, Isogai Y, Kojima T, Hirabayashi T, et al. The adipocyte-inducible secreted phospholipases PLA2G5 and PLA2G2E play distinct roles in obesity. Cell Metab. 2014;20:119–32.
Garces F, Lopez F, Nino C, Fernandez A, Chacin L, Hurt-Camejo E, et al. High plasma phospholipase A2 activity, inflammation markers, and LDL alterations in obesity with or without type 2 diabetes. Obesity (Silver Spring). 2010;18:2023–9.
Hui DY. Phospholipase a(2) enzymes in metabolic and cardiovascular diseases. Curr Opin Lipidol. 2012;23:235–40.
Wolf G. Adipose-specific phospholipase as regulator of adiposity. Nutr Rev. 2009;67:551–4.
Baltatzi M, Hatzitolios A, Tziomalos K, Iliadis F, Zamboulis C. Neuropeptide Y and alpha-melanocyte-stimulating hormone: interaction in obesity and possible role in the development of hypertension. Int J Clin Pract. 2008;62:1432–40.
Jeanrenaud B, Rohner-Jeanrenaud F. Effects of neuropeptides and leptin on nutrient partitioning: dysregulations in obesity. Annu Rev Med. 2001;52:339–51.
Arora S. Anubhuti. Role of neuropeptides in appetite regulation and obesity--a review. Neuropeptides. 2006;40:375–401.
Perez-Verdaguer M, Capera J, Serrano-Novillo C, Estadella I, Sastre D, Felipe A. The voltage-gated potassium channel Kv1.3 is a promising multitherapeutic target against human pathologies. Expert Opin Ther Targets. 2016;20:577–91.
Xu J, Koni PA, Wang P, Li G, Kaczmarek L, Wu Y, et al. The voltage-gated potassium channel Kv1.3 regulates energy homeostasis and body weight. Hum Mol Genet. 2003;12:551–9.
Thiele TE, Navarro M, Sparta DR, Fee JR, Knapp DJ, Cubero I. Alcoholism and obesity: overlapping neuropeptide pathways? Neuropeptides. 2003;37:321–37.
Grucza RA, Krueger RF, Racette SB, Norberg KE, Hipp PR, Bierut LJ. The emerging link between alcoholism risk and obesity in the United States. Arch Gen Psychiatry. 2010;67:1301–8.
Wang L, Liu X, Luo X, Zeng M, Zuo L, Wang KS. Genetic variants in the fat mass- and obesity-associated (FTO) gene are associated with alcohol dependence. J Mol Neurosci. 2013;51:416–24.
Wang KS, Zuo L, Pan Y, Xie C, Luo X. Genetic variants in the CPNE5 gene are associated with alcohol dependence and obesity in Caucasian populations. J Psychiatr Res. 2015;71:1–7.
Adrielle Lima Vieira R, Nascimento de Freitas R, Volp AC. Adhesion molecules and chemokines; relation to anthropometric, body composition, biochemical and dietary variables. Nutr Hosp. 2014;30:223–36.
Christoph MJ, Allison MA, Pankow JS, Decker PA, Kirsch PS, Tsai MY, et al. Impact of adiposity on cellular adhesion: the multi-ethnic study of atherosclerosis (MESA). Obesity (Silver Spring). 2016;24:223–30.
Fang P, Yu M, Guo L, Bo P, Zhang Z, Shi M. Galanin and its receptors: a novel strategy for appetite control and obesity therapy. Peptides. 2012;36:331–9.
Leibowitz SF. Regulation and effects of hypothalamic galanin: relation to dietary fat, alcohol ingestion, circulating lipids and energy homeostasis. Neuropeptides. 2005;39:327–32.
Gundlach AL. Galanin/GALP and galanin receptors: role in central control of feeding, body weight/obesity and reproduction? Eur J Pharmacol. 2002;440:255–68.
Kypreos KE, Karagiannides I, Fotiadou EH, Karavia EA, Brinkmeier MS, Giakoumi SM, et al. Mechanisms of obesity and related pathologies: role of apolipoprotein E in the development of obesity. FEBS J. 2009;276:5720–8.
Srivastava A, Mittal B, Prakash J, Srivastava P, Srivastava N. Analysis of MC4R rs17782313, POMC rs1042571, APOE-Hha1 and AGRP rs3412352 genetic variants with susceptibility to obesity risk in north Indians. Ann Hum Biol. 2016;43:285–8.
Breitling C, Gross A, Buttner P, Weise S, Schleinitz D, Kiess W, et al. Genetic contribution of variants near SORT1 and APOE on LDL cholesterol independent of obesity in children. PLoS One. 2015;10:e0138064.
Li YH, Liu L. Apolipoprotein E synthesized by adipocyte and apolipoprotein E carried on lipoproteins modulate adipocyte triglyceride content. Lipids Health Dis. 2014;13:136.
Wu CL, Zhao SP, Yu BL. Intracellular role of exchangeable apolipoproteins in energy homeostasis, obesity and non-alcoholic fatty liver disease. Biol Rev Camb Philos Soc. 2015;90:367–76.
Ramasamy I. Update on the molecular biology of dyslipidemias. Clin Chim Acta. 2016;454:143–85.
Kei AA, Filippatos TD, Tsimihodimos V, Elisaf MS. A review of the role of apolipoprotein C-II in lipoprotein metabolism and cardiovascular disease. Metabolism. 2012;61:906–21.
Vila G, Grimm G, Resl M, Heinisch B, Einwallner E, Esterbauer H, et al. B-type natriuretic peptide modulates ghrelin, hunger, and satiety in healthy men. Diabetes. 2012;61:2592–6.
Gruden G, Landi A, Bruno G. Natriuretic peptides, heart, and adipose tissue: new findings and future developments for diabetes research. Diabetes Care. 2014;37:2899–908.
Palmer BF, Clegg DJ. An emerging role of Natriuretic peptides: igniting the fat furnace to fuel and warm the heart. Mayo Clin Proc. 2015;90:1666–78.
Schlueter N, de Sterke A, Willmes DM, Spranger J, Jordan J, Birkenfeld AL. Metabolic actions of natriuretic peptides and therapeutic potential in the metabolic syndrome. Pharmacol Ther. 2014;144:12–27.
Tu W, Wagner EK, Eckert GJ, Yu Z, Hannon T, Pratt JH, et al. Associations between menarche-related genetic variants and pubertal growth in male and female adolescents. J Adolesc Health. 2015;56:66–72.
Kwon S, Kim D, Rhee JW, Park JA, Kim DW, Kim DS, et al. ASB9 interacts with ubiquitous mitochondrial creatine kinase and inhibits mitochondrial function. BMC Biol. 2010;8:23.
Debrincat MA, Zhang JG, Willson TA, Silke J, Connolly LM, Simpson RJ, et al. Ankyrin repeat and suppressors of cytokine signaling box protein asb-9 targets creatine kinase B for degradation. J Biol Chem. 2007;282:4728–37.
Sun G, Ukkola O, Rankinen T, Joanisse DR, Bouchard C. Skeletal muscle characteristics predict body fat gain in response to overfeeding in never-obese young men. Metabolism. 2002;51:451–6.
Oudman I, Clark JF, Brewster LM. The effect of the creatine analogue beta-guanidinopropionic acid on energy metabolism: a systematic review. PLoS One. 2013;8:e52879.
Haan YC, van Montfrans GA, Brewster LM. The high creatine kinase phenotype is hypertension- and obesity-prone. J Clin Hypertens (Greenwich). 2015;17:322.
Haan YC, Oudman I, Diemer FS, Karamat FA, van Valkengoed IG, van Montfrans GA, et al. Creatine kinase as a marker of obesity in a multi-ethnic population. Mol Cell Endocrinol. 2017;442:24–31.
Ahmed M, Gaffen SL. IL-17 in obesity and adipogenesis. Cytokine Growth Factor Rev. 2010;21:449–53.
Zizza A, Guido M, Grima P. Interleukin-17 regulates visceral obesity in HIV-1-infected patients. HIV Med. 2012;13:574–7.
Wang N, Lan D, Chen W, Matsuura F, Tall AR. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc Natl Acad Sci U S A. 2004;101:9774–9.
Kusuhara H, Sugiyama Y. ATP-binding cassette, subfamily G (ABCG family). Pflugers Arch. 2007;453:735–44.
Mitrou P, Raptis SA, Dimitriadis G. Insulin action in morbid obesity: a focus on muscle and adipose tissue. Hormones (Athens). 2013;12:201–13.
Ye J. Mechanisms of insulin resistance in obesity. Front Med. 2013;7:14–24.
Andres MA, Baptista NC, Efird JT, Ogata KK, Bellinger FP, Zeyda T. Depletion of SK1 channel subunits leads to constitutive insulin secretion. FEBS Lett. 2009;583:369–76.
Laforenza U, Scaffino MF, Gastaldi G. Aquaporin-10 represents an alternative pathway for glycerol efflux from human adipocytes. PLoS One. 2013;8:e54474.
Madeira A, Moura TF, Soveral G. Aquaglyceroporins: implications in adipose biology and obesity. Cell Mol Life Sci. 2015;72:759–71.
Mendez-Gimenez L, Rodriguez A, Balaguer I, Fruhbeck G. Role of aquaglyceroporins and caveolins in energy and metabolic homeostasis. Mol Cell Endocrinol. 2014;397:78–92.
Acknowledgements
Not applicable.
Funding
This study was supported by the grants from the Natural Science Foundation of China (81773506), the Entrepreneurial Innovation Talents Project of Qingdao City (13-CX-3), and the EFSD/CDS/Lilly Programme award (2013).
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
DFZ and WJW contributed to the conception and design. HPD and CSX organized the collection of samples and phenotypes. WJW and WJJ contributed to sample data and sequencing data management. QHT and SXL analyzed the sequencing data and WJW, YLW, and LH interpreted the analysis results. WJW, DFZ and WJJ drafted the manuscript, HPD, YLW, and CSX were involved in the discussion, and QHT, SXL, DFZ, and LH revised it. All the authors read the manuscript and gave the final approval of the version to be published. All the authors agreed to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All participants provided written informed consent for participating in the study which was approved by the local ethics committee at Qingdao CDC, Qingdao, China.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Table S1.
Summary of the sequencing reads and the mapped results for the 7 monozygotic twin pairs (DOCX 17 kb)
Additional file 2: Table S2.
The summary of differentially expressed genes with a trend of up-regulation in BMI-discordant monozygotic twin pairs and the corresponding functional annotation results (XLSX 23 kb)
Additional file 3: Table S3.
Module assignments for genes in network of WGCNA (XLSX 302 kb)
Additional file 4: Table S4.
The result for functional annotation within databases of NR, COG, KOG, KEGG, and GO for genes clustered in coral1 module (XLSX 24 kb)
Additional file 5: Table S5.
GO classification of genes in coral1 module (XLSX 13 kb)
Additional file 6: Table S6.
KEGG categories of genes in coral1 module (XLSX 12 kb)
Additional file 7: Table S7.
COG function classification in coral1 module (XLSX 12 kb)
Additional file 8: Table S8.
The hub genes found by the criterion of BMI based GS > 0.2 and MM > 0.8 with a threshold of P-value <0.05 in coral1 module (DOCX 16 kb)
Additional file 9: Figure S1.
GO classification in saddlebrown module. Annotation statistics of genes in the secondary node of GO. The horizontal axis shows secondary nodes of three categories in GO. The vertical axis displays the percentage of annotated genes versus the total gene number. The left columns display annotation information of the total genes and the right columns represent annotation information of the genes clustered in saddlebrown module. (TIFF 777 kb)
Additional file 10: Table S9.
GO classification of genes in saddlebrown module (XLSX 14 kb)
Additional file 11: Table S10.
KEGG categories of genes in saddlebrown module (XLSX 18 kb)
Additional file 12: Table S11.
COG function classification in saddlebrown module (XLSX 15 kb)
Additional file 13: Figure S2.
Scatterplots of BMI based gene significance (GS) versus module membership (MM) in the saddlebrown module. GS for BMI and MM exhibit a very significant correlation, implying that the most important (central) elements of saddlebrown module also tend to be highly correlated with BMI trait (TIFF 168 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, W., Jiang, W., Hou, L. et al. Weighted gene co-expression network analysis of expression data of monozygotic twins identifies specific modules and hub genes related to BMI. BMC Genomics 18, 872 (2017). https://doi.org/10.1186/s12864-017-4257-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-017-4257-6