
Abstract
Background
Salamanders regenerate their limbs after amputation. However, the molecular mechanism of this unique regeneration remains unclear. In this study, isobaric tags for relative and absolute quantification (iTRAQ) coupled with liquid chromatography tandem mass spectrometry (LC-MS/MS) was employed to quantitatively identify differentially expressed proteins in regenerating limbs 3, 7, 14, 30 and 42 days post amputation (dpa).
Results
Of 2636 proteins detected in total, 253 proteins were differentially expressed during different regeneration stages. Among these proteins, Asporin, Cadherin-13, Keratin, Collagen alpha-1(XI) and Titin were down-regulated. CAPG, Coronin-1A, AnnexinA1, Cathepsin B were up-regulated compared with the control. The identified proteins were further analyzed to obtain information about their expression patterns and functions in limb regeneration. Functional analysis indicated that the differentially expressed proteins were associated with wound healing, immune response, cellular process, metabolism and binding.
Conclusions
This work indicated that significant proteome alternations occurred during salamander limb regeneration. The results may provide fundamental knowledge to understand the mechanism of limb regeneration.
Similar content being viewed by others

Background
Some urodele amphibians possess the extraordinary capacity to regenerate their damaged body parts, including limbs and tails [1], jaw [2], spinal cord [3], lens [4, 5], retina [6], neuron [7] and heart [8]. For example, the missing portion of a salamander limb could be perfectly regenerated after amputation between the shoulder and hand.
Salamander limb regeneration could be divided into three major stages. The first stage is the covering of the wound. Epidermal cells around the stump migrate rapidly into the wound area and form an epithelial structure called the wound epidermis (WE) within 1 day after amputation [9]. During the second stage, cells surrounding the injury site begin to dedifferentiate, proliferate and form a mesenchymal growth zone known as the blastema. Simultaneously, the WE thickens apically to form an apical epidermal cap (AEC) [10]. In the last stage, cells in the blastema undergo patterned differentiation to generate diverse tissue structures and replicate the amputated limb parts [11]. The blastema is a heterogeneous collection of mesenchymal progenitor cells originated from mature limb tissues, including the dermis, muscle, cartilage and connective tissue underneath WE. Importantly, cells from specific tissue produce progenitor cells with restricted differentiation potential. The dermis makes epidermis, cartilage and tendons, but not muscle or Schwann cells, and muscle does not make cartilage or epidermis. Furthermore, cartilage-derived blastema cells possess positional identity, whereas Schwann-derived cells do not [11].
Regeneration of limbs is a complex process involving the activation of a number of biological processes, signaling pathways and large-scale tissue remodeling [12,13,14,15,16,17]. Understanding the mechanisms underlying in limb regeneration offers important insights into the possibilities for regenerating complex structures in adult vertebrates. Great efforts have been made to identify expression profiles and the functional role of specific genes during limb regeneration [18,19,20].
Strategies of comparative proteome have been employed to reveal substantial changes in the proteome composition and identify critical molecular during salamander limb regeneration. Rao et al. [21] identified 309 proteins that had significant changes relative to controls during blastema formation with label-free liquid chromatography/mass spectrometry. These proteins were involved in various biological process, including signaling, Ca2+ binding and translocation, transcription, translation and cell cycle. The expression of ecotropic viral integrative factor 5 (EVI5), a cell cycle-related oncoprotein, was exceptionally high throughout blastema formation. Geng et al. [22] revealed 212 proteins that participated in the differentiation of skin cells, myocytes, neurocytes, chondrocytes and osteocytes during limb regeneration, via two-dimensional fluorescence difference gel electrophoresis (2D–DIGE) and mass spectrum (MS) analyses. The combination of isobaric tagging for relative and absolute protein quantification (iTRAQ), LC-MS/MS and protein Mascot search engine is a powerful method to quantitatively identify differentially expressed proteins (DEPs) with high accuracy and reproducibility [23, 24]. In this study, we employed this method to screen proteome alterations in regenerating limbs 3, 7, 14, 30 and 42dpa. We detected 2636 proteins in total, and 253 proteins were differentially expressed during different regeneration stages. Functional bioinformatics analysis suggested that these proteins involved in various biological process categories, including wound healing and cell differentiation.
Results
Gross inspection and histological observation of regenerating limb
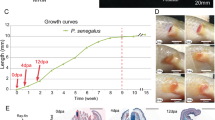
Figure 1 presents the integrated morphological and histological staging scheme of salamander limb regeneration used in the current experiment. Histological observation of regenerating limbs in the longitudinal section showed that the wound stump was covered by epidermis and gland cells. The wound epidermis lacked a basement membrane and directly contacted the underlying mesenchymal tissues at 3dpa. Muscle fragments, cellular debris and lymphocytes could be observed under the WE (Fig. 1a). At 7dpa, the WE thickened and formed an AEC. Cells from the surrounding dermal tissue, muscle (M) and bone (B) dedifferentiated and accumulated under the AEC (Fig. 1b). By 14dpa, the amount of undifferentiated mesenchymal cells increased and formed a blastema (Fig. 1c). By 30dpa, the blastema developed to form an early limb bud (Fig. 1d). At 42dpa, limb with initial digits was regenerated (Fig. 1e).

The morphological and histological appearance at different days post amputation. Gross inspection of Cynops orientalis limb regeneration (left). Samples were amputated 1 mm over the elbow at different days post-amputation. HE staining of longitudinal sections of regenerating limbs at different days post amputation (right). a Section at 3dpa. Wound stump covered by epidermis and gland cells without basement membrane. b Section at 7dpa. Undifferentiated mesenchymal cells accumulating under the thickened AEC (M: muscle, B: bone). c Section at 14dpa. Formation of blastema. d Section at 30dpa. Formation of initial limb bud. e Section of 42dpa. Regenerating limb with initial digits
Protein identification and quantification
Tryptic peptides were labeled with iTRAQ tags, and the analytical separation and identification of the samples for each biological replicate were performed by LC-MS/MS. A total of 6042 unique peptides were detected, and 2636 proteins with a mascot score of at least 13 were identified between the experimental and control groups. (Additional file 1: Table S1).
Differentially expressed proteins analysis
The relative quantification of proteins was based on the ratio of the peak areas from the MS/MS spectra. Among them, 590 proteins had quantitative information. In the current experiment, proteins with iTRAQ ratios below the low range (0.8) were considered to be down-regulated, whereas those above the high range (1.2) were considered to be up-regulated. The identified DEPs were also detected in at least two of the three biological replicates. Based on these criteria, we identified 253 proteins with a significant difference among the regenerating groups compared with the control group (Fig. 2 and Additional file 2: Table S2). Our findings showed that the number of DEPs peaked in the temporal comparison of 30dpa vs. control, 42dpa vs. control, and 7dpa vs. control.

Numbers of DEPs in the regenerated limb tissues compared with the control group
Among these DEPs, 153 DEPs were up-regulated (Additional file 3: Figure S1A, Additional file 2: Table S2) and Myeloperoxidase (MPO) was the most up-regulated protein (3dpa vs. control ratio; mean 5.491). The up-regulated proteins were involved in transport and metabolism, posttranslational modification, transcription, cytoskeleton, such as macrophage-capping protein (CAPG), coronin-1A (CORO1A), annexinA1 (ANXA1), hemopexin, cathepsin B (CTSB), and eukaryotic translation initiation factor 5A isoform II (EIF5A2). In total, 87 DEPs were down-regulated (Additional file 3: Figure S1B, Additional file 2: Table S2) and involved in transcription, signal transduction mechanisms, transport and metabolism. The down-regulated DEPs included extracellular matrix organization-related proteins, such as keratin, type II cytoskeletal, annexinA 2-A(anxa2-a), matrilin-4(MATN4), cadherin-13 (CDH13), basement membrane-specific heparan sulfate proteoglycan core protein (HSPG2), and collagen alpha-1(XI) chain (COL11A1). Keratin was the most down-regulated protein (3dpa vs. control ratio; mean 0.06633). A total of 13 DEPs were identified that exhibited a variation trend as a wave change post amputation between the experimental and control groups.
Function and bioinformatics analyses
Based on Gene Ontological (GO) analysis of DEPs, there’re 200 proteins which have GO annotations and can be categorized functionally using WEGO (Fig. 3). A protein was counted more than once when the protein was assigned to more than one category. Figure 3 stratifies the proteins according to biological process, cellular component and molecular function. The DEPs were grouped into 23 biological process categories: cellular processes (83.6%), metabolic processes (70.6%), biological regulation (51.2%), pigmentation (43.3%), multicellular organismal processes (40.8%), developmental processes (37.8%), cellular component organization (36.3%), localization (34.8%), establishment of localization (30.8%), response to stimulus (28.4%), anatomical structure formation (16.9%), cellular component biogenesis (16.4%), immune system processes (12.4%), death (10.0%), reproduction (8.5%), reproductive process (8.5%), viral reproduction (7.5%), multi-organism process (6.5%), biological adhesion (4.5%), growth (3.0%), locomotion (4.0%), rhythmic process (0.5%) and cell killing (0.5%). Important molecular functions associated with the datasets include binding, catalytic activity, and structural molecule activity. A detailed GO annotation of the differentially expressed proteins was provided in Additional file 4: Table S3.

GO distribution analysis of differentially expressed proteins during regeneration from the control and treatment groups (3, 7, 14, 30 and 42dpa). In total, 200 differentially expressed proteins were categorized based on “Cellular Component”, “Molecular Function”, and “Biological Process” using WEGO
Those differentially expressed proteins with significant changes were profiled based on their COG functional classification and categorized into as many as 18 COGs (Fig. 4). “General function prediction only” represented the largest group, followed by “Posttranslational modification, protein turnover, chaperones” and “translation, ribosomal structure and biogenesis”.

COG classification of differentially expression proteins in iTRAQ data. The X-axis represents the COG categories, whereas the Y-axis represents protein quantity. The picture displays the statistics as the number of different functional proteins in the sample
To identify the biological pathways involved in limb regeneration, we mapped the annotated sequences to the reference canonical pathways in the Kyoto Encyclopedia of Genes and Genomes (KEGG). There’re 232 KEGG pathway annotated by all these DEPs. (Additional file 5: Table S4). When the FDR-corrected P-value was set as <0.05, 29 KEGG pathways were identified to be significant (Table 1).
To illustrate changes in biological processes, protein expression data were mapped onto the KEGG Mapper-Search & Color Pathway (http://www.genome.jp/kegg/tool/map_pathway2.html) and changes in expression are indicated by color (Fig. 5). PI3k-Akt was the most enrichment pathway which associated with nine DEPs. IgA1 immunoglobulin, Collagen alpha-1(II) chain (COL2A1), Collagen alpha-3(VI) chain (CO6A3), Collagen alpha-1(XI) chain (COL11A1) and ITGB4 were down-regulated during regeneration, whereas heat shock protein HSP 90-alpha (HSP90AA1), Hemopexin (HPX), and Mitogen-activated protein kinase 3(MAPK3) were up-regulated. Serine/threonine-protein phosphatase2A regulatory subunit A (PPP2R1A) showed no obvious changes. Three genes (HPX, COL2A1 and ITGB4) involved in this pathway were randomly selected for qRT-PCR, the result was consistent with that of iTRAQ assay (Additional file 6: Figure S2).

Protein expression mapped to the KEGG pathway. The protein expression profiles mapped to the PI3k-Akt signaling pathway. DEPs are marked with different colors. The red color indicates up-regulated, blue color indicates down-regulated, and pink indicates no obvious change
PPI network construction
The STRING database aims to provide a global perspective for as many organisms as is feasible. To investigate the interactions among regeneration-related proteins, DEPs predicatively related to wound healing and the immune response, cell proliferation and differentiation were integrated with information from the STRING database to construct a PPI network (Additional file 7: Table S5).
A network was generated based on the protein–protein interactions of the 27 wound healing and immune response-related proteins in Fig. 6a. Only RHOA was located in the center of the network, indicating that proteins in the Xenopus Silurana database were limited.

Interaction networks of wound healing and the immune response (a), cell proliferation and cell differentiation-related DEPs (b) identified by the iTRAQ experiment or Text Mining databases. The light blue lines represent data from curated databases. The purple lines represent experimentally determined data, and the yellow lines represent textmining. The black lines represent co-expression. The green lines represent the gene neighborhood, and the blue lines represent curated databases
CTNNB1 and RHOA were the key proteins for cell proliferation and cell differentiation in the PPI network. In Fig. 6b, an interaction network including STAT5, RHOA, PCNA, NME2, SHMT2, CTNNB1, HMOX1, VASP and CUL3 was generated, whereas no interactions were observed among CDH13, CALR, CORO1A, STIP, G6PD, Serpinb5 and EVPL.
Validation of differentially expressed proteins by qRT-PCR analysis
To provide further information regarding the correspondence between proteins and their mRNA expression patterns, quantitative real-time polymerase chain reaction (qRT-PCR) was performed to investigate the dynamic transcriptional expression patterns of nine representative DEPs. The expression patterns determined by qRT-PCR were consistent with those obtained by iTRAQ, with 80% agreement between the qRT-PCR and iTRAQ results (Fig. 7). This result indicated that the differential proteomic analysis results in this study were reliable.

Quantitative RT-PCR analyses of gene expression in regenerating limb. Quantitative expression of genes for the regenerating time point calculated based on Ct value normalized against the housekeeping Beta-actin gene
Discussion
iTRAQ combined with LC–MS is one of the most powerful methodologies in quantitative proteomics [25, 26]. In the present study, we employed this technique to study protein expression profiles during limb regeneration to identify proteins that are critical for regeneration. Our findings provide primary proteomic information during limb regeneration that may be useful in understanding the mechanisms of limb regeneration. We detected a total of 6042 peptides and 2636 proteins in all samples (0, 3, 7, 14, 30 and 42dpa), whereas Rao et al. [21] detected 1624 peptides in all of their samples (0, 1, 4, and 7dpa). However, some proteins that were previously reported to be critical for limb regeneration were not identified in our study or Raos’ study, such as Fgf-8, Hox, sonic hedgehog, prod1 and nAG. This difference may due to an inability of the current LC-MS/MS technology to sensitively detect low abundance proteins.
The grouping of the proteins into clusters combined with functional classification analysis provided clear evidence for the involvement of different characteristic temporal patterns of protein expression during Cynops orientalis limb regeneration. Our results confirmed a number of earlier studies on signaling, cytoskeletal, ECM and metabolism changes.
Wound healing stage
Fourteen DEPs for wound response and healing post amputation were detected in the current experiment (Additional file 7: Table S5), including RHOA, annexinA2, complement factor B, complement C3, coagulation factor XIII, and Signal transducer activator of transcription 5B (STAT5B). RHOA links plasma membrane receptors to the assembly of focal adhesions and actin stress fibers and plays an essential role for the apical junction formation of keratinocyte cell-cell adhesion. Previous study also validated the association of annexin A2 down-regulation for cell proliferation and differentiation in the mesenchymal progenitor cells of regenerating limbs [22].
Skin wounds in adult mammals heal after the re-epithelization on the formed granulation tissue and often result in scars or keloids. In salamander, the wound epithelium is formed quickly and then thickens to form the AEC. A featured histological appearance of AEC is the absence of the basal membrane, which enables the direct interaction among the epithelium, nerves, and mesenchymal tissue. Coincident with this histological appearance, the cell density was much higher at the amputation plane, indicating that the blastema forms primarily by the distal migration and accumulation of dedifferentiated cells under the wound epidermis [9]. Transplantation of full thickness skin graft onto the stump will terminate the regeneration process. In the current experiment, we detected the upregulation of Villin-1 and epidermal differentiation-specific protein, which could accelerate the formation of the wound epithelium. Moreover, the expression of basement membrane-specific heparan sulfate proteoglycan core protein, which is a major component of the basal membrane, was down-regulated.
The immune system is critical for the body’s defense against exogenous pathogens. Previous histological studies have indicated the presence of lymphocytes and macrophages in early blastema after limb amputation [21, 27]. In recent years, there has been a growing appreciation of the contribution of cellular components of the immune system to regeneration. Macrophages are professional phagocytes of the system. Regulation of the immune milieu has been suggested to be a critical precondition for adult regeneration. It was reported that systemic macrophage depletion in salamander may result in permanent failure of limb regeneration associated with extensive fibrosis and disregulation of extracellular matrix component gene expression [28]. In a recent study, Cattin and Burden et al. demonstrated that macrophages are necessary for the bridge of the migrating cords of Schwann cells during nerve damage [29]. We identified 16 proteins related to the immune response in the current study (Additional file 7: Table S5). Among them, 4 proteins were closely associated with macrophage function, including Coronin-1A, Macrophage-capping protein, Complement factor B and Complement C3. Coronin-1A, an actin binding protein that is involved in the regulation of the immune system [30, 31], was up-regulated at 7dpa and lasting until 42dpa. Macrophage-capping protein, a gelsolin family actin-regulatory protein involved in cell signaling, phagocytosis and motility [32], was significantly up-regulated at 3dpa and 7dpa. These results further implied the critical role of macrophages during limb regeneration. An in-depth understanding of the role of immune systems on regeneration may provide clues to therapeutic avenues to restore damaged tissue in mammals.
Blastema formation
The major signal transduction pathways identified during regeneration included FGF, Wnt, Hedgehog (Hh), retinoic acid (RA), Notch, BMP, IGF, PI3K-AKT and MAPK signaling pathways. The Wnt pathway is essential for the proliferation and maintenance of stem cells, and is involved in the initial step of limbs morphogenesis and regeneration. Yokoyama H et al. demonstrated that inhibition of Wnt/β-catenin signaling could inhibit limb regeneration during the early stage [33]. In the current experiment, carboxypeptidaseZ (CPZ), a secreted Zn-dependent enzyme that is enriched in the extracellular matrix [34, 35], was up-regulated at 7, 14, 30 and 42dpa. MAPK3, which regulates various cellular processes such as cell proliferation, cycle progression, response to extracellular signals, was found up-regulated at 14dpa.
Nerves play a critical role in tissue repair and regeneration. In the case of salamander limb regeneration, regenerating axons are key elements and denervation may inhibit the ignition of blastemal formation. This effect is mediated by the interaction of nerves with the WE. In the current study, we detected two up-regulated proteins that are closely related to axon growth. Proactivatorpoly peptide (PSAP) is the precursor of sphingolipid activator proteins and is abundant in the nervous system and muscle, which supplied neurotrophic effects in vivo or in vitro [36]. RHOA is a member of the Rho family of small GTPases, which regulates cytoskeletal dynamics, transcription, cell cycle progression and cell transformation [37]. Peripheral nerve injury triggers the activation of RHOA in spinal motor and peripheral sensory neurons [38].
Cell differentiation and limb regeneration
Cells adjacent to the amputation site were freed by protease-induced histolysis (Proteasome activator complex and Cathepsin B, as found in the current experiment) and dedifferentiated. Dedifferentiated cell were characterized by the expression of genes associated with the dedifferentiated state, including msx1, Nrad, Notch, Klf4, Sox2, c-myc and LIN28 [15, 18, 39]. It has been suggested that apoptotic pathways are closely related to cell dedifferentiation [40]. In this study, nucleoside diphosphate kinase B, a member of the nm23 family and inhibitor of c-myc transcription, was found down-regulated.
Cell differentiation is crucial for limb regeneration, and cells in the blastema may undergo proliferation and differentiation to generate the missing structures of limb, such as skin, muscle, bone, cartilage, nerve and blood vessels. During this stage, regenerating tissue exhibited active biosynthetic activity, and 28 proteins were found associated with cell differentiation (Additional file 7: Table S5). In addition, 26 and 18 proteins involved in transcription and translation were detected (Additional file 7: Table S5). For example, calreticulin (CALR) was significantly up-regulated at 14 and 30dpa. Eukaryotic translation initiation factor 5A-2 was significantly up-regulated at all time points.
Analysis of the protein expression profile is useful to identify the initial molecules and mechanisms triggering limb regeneration and the difference in the wound healing pattern between salamander and mammals. Mimicking the mechanisms of limb regeneration in salamander may improve regenerative ability in humans via gene therapy, cell therapy and chemical therapy.
Conclusions
In this study, an iTRAQ-based quantitative proteomic approach was applied to detect the overall proteome changes during Cynops orientalis limb regeneration. The results indicated that significant proteomic alternations occurred during the process. Totally 253 proteins were found differentially expressed and involved in 29 KEGG pathways. The DEPs detected may provide fundamental basis to understand the mechanism of limb regeneration. Several proteins, including coronin, RHOA and Macrophage-capping protein were first found differentially expressed, and the role of these proteins should be fully investigated in future study.
Methods
Sample preparation
Cynops orientalis were purchased from a pet market. All surgical procedures were performed under general anesthesia with 0.1% MS-222 (ethyl-p-aminobenzoate, Sigma-Aldrich, Steinheim, Germany) in a 1-L tap water-bath. The right forelimb of each animal was amputated 1 mm over elbow, and limb tissues were collected 3, 7, 14, 30 and 42dpa by cutting the arm 1 mm from the first amputation. The tissue removed distal to the amputation site acted as the control. The collected tissues were rinsed twice in filtered water and twice in 1× phosphate-buffered saline (PBS) and were then frozen in liquid nitrogen or stored at −80 °C until they were prepared for extraction.
Histology
The tissues mentioned above were fixed in a PBS buffered formaldehyde solution for 48 h. Decalcification with EDTA was performed for 1 to 2 months. The fixed tissue samples were dehydrated in a graded series of alcohol, followed by xylene. Then, the samples were infiltrated overnight with paraffin. The tissues were then sectioned at 8 μm. Hematoxylin and eosin (HE) staining was performed.
Protein extraction, iTRAQ labeling and SCX fractionation
Tissue samples from each group were ground into powder in liquid nitrogen, and a protein preparation was performed as described by Li et al. [41]. Three biological replicates from each group were used to prepare protein samples. The process of iTRAQ labeling and SCX fractionation was performed as described by Josselin et al. [25, 26]. Briefly, one hundred micrograms of protein sample was digested with Trypsin Gold (Promega, Madison, WI, USA) and labeled according to the manufacturer’s protocol for 8-plex iTRAQ reagent (Applied Biosystems). Samples were labeled with the iTRAQ tags as follow: control (113 tag), sample 3dpa (118 tag), sample 7dpa (121 tag), sample 14dpa (119 tag), sample 30dpa (117 tag), and sample 42dpa (115 tag). SCX chromatography was performed on a LC-20AB HPLC Pump system (Shimadzu, Kyoto, Japan) with a 4.6 × 250 mm2 Ultremex SCX analytical column. The labeled digests were subjected to LC-MS/MS analysis.
LC-ESI-MS/MS analysis based on triple TOF 5600
Each fraction was resuspended in buffer A (5% ACN, 0.1%FA) and centrifuged at 20000 g for 10 min to achieve a final concentration of approximately 0.5 μg/μl. Then, 10 μl of supernatant was loaded on a LC-20 AD nanoHPLC (Shimadzu, Kyoto, Japan) by the auto sampler onto a 2-cm C18 trap column. Then, the peptides were eluted onto a 10-cm analytical C18 column packed in-house. The samples were loaded at 8 μL/min for 4 min. Then, the 35-min gradient was run as follows: 300 nL/min starting from 2 to 35% B (95%ACN, 0.1%FA); 5 min linear gradient to 60%, 2 min linear gradient to 80%, maintenance at 80% B for 4 min, and return to 5% in 1 min.
Data acquisition was performed with a TripleTOF 5600 System (AB SCIEX, Concord, ON) fitted with a Nanospray III source (AB SCIEX, Concord, ON), and a pulled quartz tip served as the emitter (New Objectives, Woburn, MA). Data were acquired using an ion spray voltage of 2.5 kV, curtain gas of 30 psi, nebulizer gas of 15 psi, and an interface heater temperature of 150. The MS was operated with a RP of greater than or equal to 30,000 FWHM for TOF MS scans. For IDA, survey scans were acquired over 250 ms, and up to 30 product ion scans were collected; a threshold of 120 counts per second (counts/s) was exceeded with a 2+ to 5+ charge-state. The total cycle time was fixed to 3.3 s. The Q2 transmission window was 100 Da for 100%. Four time bins were summed for each scan at a pulser frequency value of 11 kHz by monitoring the 40 GHz multichannel TDC detector with a four-anode channel to detect ions. A sweeping collision energy setting of 35 ± 5 eV coupled with iTRAQ adjust rolling collision energy was applied to all precursor ions for collision-induced dissociation. Dynamic exclusion was set at 1/2 of the peak width (15 s), and then, the precursor was refreshed off the exclusion list.
iTRAQ data analysis
The MS data were processed by Proteome Discoverer software (version 1.2.0.208) (Thermo Scientific, Bremen, Germany), and protein identification and quantification were performed using the Mascot search engine (Matrix Science, London, UK; version 2.3.02) against the Salamandridae database containing 134,205 sequences.
For protein identification, a mass tolerance of 0.05 Da was permitted for intact peptide masses and 0.1 Da for fragmented ions, with an allowance for one missed cleavage in the trypsin digests. Gln- > pyro-Glu (N-term Q), oxidation (M), and deamination (NQ) were potential variable modifications. Carbamidomethyl (C), iTRAQ8plex (N-term), and iTRAQ 8plex (K) were fixed modifications. The charge states of peptides were set to +2 and +3. Specifically, an automatic decoy database search was performed in Mascot by choosing the decoy checkbox in which a random database sequence was generated and tested for raw spectra as well as the real database. To reduce the probability of false peptide identification, only peptides with significance scores (≥20) at the 99% confidence level with a Mascot probability analysis greater than “identity” were counted as identified. Each confident protein identification involved at least one unique peptide.
Analysis of differentially expressed proteins
A protein must contain at least two unique peptides for protein quantitation. The quantitative protein ratios were weighted and normalized by the median ratio in Mascot. Only ratios with p-values <0.05 and fold changes of >1.2 or <0.8 were considered to be significant in the current study.
Bioinformatics analysis
Functional annotations of the proteins were conducted with the Blast2GO program against the non-redundant protein database (NR; NCBI). The protein accessions list of native format containing corresponding GO annotations was submitted to the Web Gene Ontology Annotation Plot (WEGO: http://wego.genomics.org.cn/cgi-bin/wego/index.pl). The KEGG database (http://www.genome.jp/kegg/) and the COG database (http://www.ncbi.nlm.nih.gov/COG/)were used to classify and group these identified proteins.
To evaluate the similarities or differences of the protein expression profiles among different groups, Cluster3.0/TreeView software was used for hierarchical clustering of the identified proteins [42]. In the current study, the intensities of the identified proteins were used for hierarchical clustering, and the missing values were treated as zero.
PPI network construction
STRING v10.1 (http://string-db.org/) was applied to analyze the protein–protein interaction (PPI) of differentially expressed protein identified in the current study and to construct PPI networks. The protein interaction information was extracted from the orthologous proteins of Xenopus Silurana. The active prediction methods were enabled, such as Database, Experiment and Text Mining [43].
Quantitative real-time PCR (qRT-PCR) analysis for gene expression
qRT-PCR was performed using SYBR Premix Ex Taq (TaKaRa, Dalian, China) and a Thermal Cycler CFX96 Real Time-PCR detection system (Bio-Rad, Hercules, CA, USA) with the following parameters: 95 °C for 30 s; 40 cycles at 95 °C for 10 s; 52 °C for 10 s; and 72 °C 10 s. The β-actin gene was used as the internal control. For qRT-PCR, total RNA was extracted from the regenerating limb using the RNAiso plus reagent (Takara, Dalian, China) following the manufacturer’s protocols. The concentration and purity of total RNA were measured using a GE Nanovue™ Spectrophotometer (GE Healthcare Biosciences, Pittsburgh, USA). cDNA was synthesized using a SYBR Prime Script™ RT Master Mix (Perfect Real Time) Kit (Takara, Dalian, China). The gene-specific primers for qRT-PCR and gene annotation are listed in Table 2. The relative expression of each gene was calculated with the 2−ΔΔCt method. There were three biological sample replicates, and each biological sample replicate included three technical replicates.
Abbreviations
- MAPK3:
-
Mitogen-activated protein kinase 3
- 3PPP2R1A:
-
Serine/threonine-protein phosphatase 2A 65 kDa regulatory subunit A alpha isoform
- CALR:
-
Calreticulin
- CAPG:
-
Macrophage-capping protein
- CDH13:
-
Cadherin-13
- COL2A:
-
Collagen alpha-1(II) chain
- CORO1A:
-
Coronin-1A
- CTNNB1:
-
Catenin beta-1
- CUL3:
-
Cullin-3
- DEPs:
-
Differentially expressed proteins
- DPA:
-
Days post amputation
- EVPL:
-
Envoplakin
- FDR:
-
False discovery rate
- G6PD:
-
Glucose-6-phosphate 1-dehydrogenase
- GO:
-
Gene Ontology
- HMOX1:
-
Heme oxygenase 1
- HPX:
-
Hemopexin
- HSP90AA1:
-
Heat shock protein HSP 90-alpha
- ITGB4:
-
Integrin beta-4
- iTRAQ:
-
isobaric tags for relative and absolute quantification
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LC-MS/MS:
-
liquid chromatography tandem mass spectrometry
- NME2:
-
Nucleoside diphosphate kinase B
- PCNA:
-
Proliferating cell nuclear antigen
- RHOA:
-
Transforming protein RhoA
- SHMT2:
-
Serine hydroxymethyltransferase,mitochondrial
- STAT5:
-
Signal transducer and activator of transcription 5B
- STIP:
-
Stress-induced-phosphoprotein 1
- VASP:
-
Vasodilator-stimulated phosphoprotein
References
Iten LE, Bryant SV. Regeneration from different levels along the tail of the newt, Notophthalmus Viridescens. J Exp Zool. 1976;196(3):293–306.
Ghosh S, Thorogood P, Ferretti P. Regenerative capability of upper and lower jaws in the newt. Int. J. Dev. Biol. 1994;38(3):479–90.
Zukor KA, Kent DT, Odelberg SJ. Fluorescent whole-mount method for visualizing three-dimensional relationships in intact and regenerating adult newt spinal cords. Dev. Dyn. 2010;239(11):3048–57.
Grogg MW, Call MK, Okamoto M, Vergara MN, Del Rio-Tsonis K, Tsonis PA. BMP inhibition-driven regulation of six-3 underlies induction of newt lens regeneration. Nature. 2005;438(7069):858–62.
Henry JJ, Tsonis PA. Molecular and cellular aspects of amphibian lens regeneration. Prog Retin Eye Res. 2010;29(6):543–55.
Sakakibara S, Hiramatsu H, Takahashi Y, Hisatomi O, Kobayashi Y, Sakami S, Saito T, Tokunaga F. Opsin expression in adult, developing, and regenerating newt retinas. Brain Res Mol Brain Res. 2002;103(1–2):28–35.
Berg DA, Kirkham M, Beljajeva A, Knapp D, Habermann B, Ryge J, Tanaka EM, Simon A. Efficient regeneration by activation of neurogenesis in homeostatically quiescent regions of the adult vertebrate brain. Development. 2010;137(24):4127–34.
Singh BN, Koyano-Nakagawa N, Garry JP, Weaver CV. Heart of newt: a recipe for regeneration. J Cardiovasc Transl Res. 2010;3(4):397–409.
Campbell LJ, Crews CM. Wound epidermis formation and function in urodele amphibian limb regeneration. Cell. Mol. Life Sci. 2008;65(1):73–9.
Christensen RN, Tassava RA. Apical epithelial cap morphology and fibronectin gene expression in regenerating axolotl limbs. Dev. Dyn. 2000;217(2):216–24.
Kragl M, Knapp D, Nacu E, Khattak S, Maden M, Epperlein HH, Tanaka EM. Cells keep a memory of their tissue origin during axolotl limb regeneration. Nature. 2009;460(7251):60–5.
Ponomareva LV, Athippozhy A, Thorson JS, Voss SR. Using Ambystoma Mexicanum (Mexican axolotl) embryos, chemical genetics, and microarray analysis to identify signaling pathways associated with tissue regeneration. Comp Biochem Physiol C Toxicol Pharmacol. 2015;178:128–35.
Levesque M, Gatien S, Finnson K, Desmeules S, Villiard E, Pilote M, Philip A, Roy S. Transforming growth factor: beta signaling is essential for limb regeneration in axolotls. PLoS One. 2007;2(11):e1227.
Monaghan JR, Maden M. Visualization of retinoic acid signaling in transgenic axolotls during limb development and regeneration. Dev Biol. 2012;368(1):63–75.
Cadinouche MZ, Liversage RA, Muller W, Tsilfidis C. Molecular cloning of the Notophthalmus Viridescens radical fringe cDNA and characterization of its expression during forelimb development and adult forelimb regeneration. Dev. Dyn. 1999;214(3):259–68.
Levesque M, Villiard E, Roy S. Skin wound healing in axolotls: a scarless process. J Exp Zool B Mol Dev Evol. 2010;314(8):684–97.
Denis JF, Levesque M, Tran SD, Camarda AJ, Roy S. Axolotl as a model to study Scarless wound healing in vertebrates: role of the transforming growth factor Beta signaling pathway. Adv. Wound Care. 2013;2(5):250–60.
Crews L, Gates PB, Brown R, Joliot A, Foley C, Brockes JP, Gann AA. Expression and activity of the newt Msx-1 gene in relation to limb regeneration. Proc. Biol. Sci. 1995;259(1355):161–71.
Grassme KS, Garza-Garcia A, Delgado JP, Godwin JW, Kumar A, Gates PB, Driscoll PC, Brockes JP. Mechanism of action of secreted newt anterior gradient protein. PLoS One. 2016;11(4):e0154176.
Bandyopadhyay A, Tsuji K, Cox K, Harfe BD, Rosen V, Tabin CJ. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2006;2(12):e216.
Rao N, Jhamb D, Milner DJ, Li B, Song F, Wang M, Voss SR, Palakal M, King MW, Saranjami B, et al. Proteomic analysis of blastema formation in regenerating axolotl limbs. BMC Biol. 2009;7:83.
Geng X, Xu T, Niu Z, Zhou X, Zhao L, Xie Z, Xue D, Zhang F, Xu C. Differential proteome analysis of the cell differentiation regulated by BCC, CRH, CXCR4, GnRH, GPCR, IL1 signaling pathways in Chinese fire-bellied newt limb regeneration. Differentiation. 2014;88(4–5):85–96.
Dayon L, Pasquarello C, Hoogland C, Sanchez JC, Scherl A. Combining low- and high-energy tandem mass spectra for optimized peptide quantification with isobaric tags. J Proteome. 2010;73(4):769–77.
Karp NA, Huber W, Sadowski PG, Charles PD, Hester SV, Lilley KS. Addressing accuracy and precision issues in iTRAQ quantitation. Mol. Cell. Proteomics. 2010;9(9):1885–97.
Unwin RD, Griffiths JR, Whetton AD. Simultaneous analysis of relative protein expression levels across multiple samples using iTRAQ isobaric tags with 2D nano LC-MS/MS. Nat Protoc. 2010;5(9):1574–82.
Gan CS, Chong PK, Pham TK, Wright PC. Technical, experimental, and biological variations in isobaric tags for relative and absolute quantitation (iTRAQ). J Proteome Res. 2007;6(2):821–7.
Young HE, Bailey CF, Markwald RR, Dalley BK. Histological analysis of limb regeneration in postmetamorphic adult Ambystoma. Anat Rec. 1985;212(2):183–94.
Godwin JW, Pinto AR, Rosenthal NA. Macrophages are required for adult salamander limb regeneration. Proc Natl Acad Sci U S A. 2013;110(23):9415–20.
Cattin AL, Burden JJ, Van Emmenis L, Mackenzie FE, Hoving JJ, Garcia Calavia N, Guo Y, McLaughlin M, Rosenberg LH, Quereda V, et al. Macrophage-induced blood vessels guide Schwann cell-mediated regeneration of peripheral nerves. Cell. 2015;162(5):1127–39.
Punwani D, Pelz B, Yu J, Arva NC, Schafernak K, Kondratowicz K, Makhija M, Puck JM. Coronin-1A: immune deficiency in humans and mice. J Clin Immunol. 2015;35(2):100–7.
Xu XJ, Tang YM. Coronin-1a is a potential therapeutic target for activated T cell-related immune disorders. APMIS. 2015;123(1):89–91.
De Corte V, Van Impe K, Bruyneel E, Boucherie C, Mareel M, Vandekerckhove J, Gettemans J. Increased importin-beta-dependent nuclear import of the actin modulating protein CapG promotes cell invasion. J Cell Sci. 2004;117(Pt 22):5283–92.
Yokoyama H, Maruoka T, Ochi H, Aruga A, Ohgo S, Ogino H, Tamura K. Different requirement for Wnt/beta-catenin signaling in limb regeneration of larval and adult Xenopus. PLoS One. 2011;6(7):e21721.
Novikova EG, Reznik SE, Varlamov O, Fricker LD. Carboxypeptidase Z is present in the regulated secretory pathway and extracellular matrix in cultured cells and in human tissues. J Biol Chem. 2000;275(7):4865–70.
Moeller C, Swindell EC, Kispert A, Eichele G. Carboxypeptidase Z (CPZ) modulates Wnt signaling and regulates the development of skeletal elements in the chicken. Development. 2003;130(21):5103–11.
Hosoda Y, Miyawaki K, Saito S, Chen J, Bing X, Terashita T, Kobayashi N, Araki N, Shimokawa T, Hamada F, et al. Distribution of prosaposin in the rat nervous system. Cell Tissue Res. 2007;330(2):197–207.
Hall A, Lalli G. Rho and Ras GTPases in axon growth, guidance, and branching. Cold Spring Harb Perspect Biol. 2010;2(2):a001818.
Leslie JR, Imai F, Zhou X, Lang RA, Zheng Y, Yoshida Y. RhoA is dispensable for axon guidance of sensory neurons in the mouse dorsal root ganglia. Front Mol Neurosci. 2012;5:67.
Shimizu-Nishikawa K, Tsuji S, Yoshizato K. Identification and characterization of newt rad (ras associated with diabetes), a gene specifically expressed in regenerating limb muscle. Dev. Dyn. 2001;220(1):74–86.
Tanaka E, Galliot B. Triggering the regeneration and tissue repair programs. Development. 2009;136(3):349–53.
Li J, Ding X, Han S, He T, Zhang H, Yang L, Yang S, Gai J. Differential proteomics analysis to identify proteins and pathways associated with male sterility of soybean using iTRAQ-based strategy. J Proteome. 2016;138:72–82.
Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95(25):14863–8.
Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39(Database issue):D561–8.
Acknowledgments
We would like to thanks LC science for analysis of mass spectrometric data.
Funding
This work was supported by the National Natural Science Foundation of China (31670996).
Availability of data and materials
The datasets generated and/or analysed during the current study are not publicly available due sequencing data has not been completely verified but are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
FC conceived and designed the project. JT analyzed and interpreted the iTRAQ data and was a major contributor in writing the manuscript. JC performed the histological examination of the regeneration limb, JT, YY, LY and MS planned and executed experiments. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All animal experimental protocols were approved by the Northwest university animal care and use committee.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Table S1.
Details for the identified proteins. (XLS 3255 kb)
Additional file 2: Table S2.
The differentially expression proteins (DEPs) identified during Cynops orientalis limb regeneration. (XLSX 194 kb)
Additional file 3: Figure S1.
Hierarchy clustering analysis of the DEPs on the limb regenerated stages of Cynops orientalis. (ZIP 670 kb)
Additional file 4: Table S3.
GO annotations of the differentially expressed proteins. (XLSX 20 kb)
Additional file 5: Table S4.
Differential expression proteins were assigned to 232 KEGG pathway. (XLSX 22 kb)
Additional file 6: Figure S2.
qRT-PCR validation of three selected genes involved in PI3k-Akt signaling pathway. (TIFF 122 kb)
Additional file 7: Table S5.
Gene ontology function enrichment analysis of DEPs involved in PPI network construction. (XLSX 15 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tang, J., Yu, Y., Zheng, H. et al. ITRAQ-based quantitative proteomic analysis of Cynops orientalis limb regeneration. BMC Genomics 18, 750 (2017). https://doi.org/10.1186/s12864-017-4125-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-017-4125-4