
Abstract
Background
Our initial genome-wide association study (GWAS) identified 20 promising candidate genes for milk fatty acid (FA) traits in a Chinese Holstein population, including PRLR, MOGAT1, MINPP1 and CHUK genes. In this study, we performed whether they had significant genetic effects on milk FA traits in Chinese Holstein.
Results
We re-sequenced the entire exons and 3000 bp of the 5′ and 3′ flanking regions, and identified 11 single nucleotide polymorphisms (SNPs), containing four in PRLR, two in MOGAT1, two in MINPP1, and three in CHUK. The SNP-based association analyses showed that all the 11 SNPs were significantly associated with at least one milk FA trait (P = 0.0456 ~ < 0.0001), and none of them had association with C11:0, C13:0, C15:0 and C16:0 (P > 0.05). By the linkage disequilibrium (LD) analyses, we found two, one, one, and one haplotype blocks in PRLR, MOGAT1, MINPP1, and CHUK, respectively, and each haplotype block was significantly associated with at least one milk FA trait (P = 0.0456 ~ < 0.0001). Further, g.38949011G > A in PRLR, and g.111599360A > G and g.111601747 T > A in MOGAT1 were predicted to alter the transcription factor binding sites (TFBSs). A missense mutation, g.39115344G > A, could change the PRLR protein structure. The g.20966385C > G of CHUK varied the binding sequences for microRNAs. Therefore, we deduced the five SNPs as the potential functional mutations.
Conclusion
In summary, we first detected the genetic effects of PRLR, MOGAT1, MINPP1 and CHUK genes on milk FA traits, and researched the potential functional mutations. These data provided the basis for further investigation on function validation of the four genes in Chinese Holstein.
Similar content being viewed by others


Background
For dairy breeding, the weight of milk components has attracted more and more attention. Fatty acids (FAs) are the important components of milk fat, and the milk fat contains 70% of saturated fatty acids (SFAs) and 30% unsaturated fatty acids (UFAs) [1]. While, the ideal nutritional milk fat contain 10% polyunsaturated fatty acids, 82% monounsaturated fatty acids and 8% SFAs, that can not be accomplished by modifying diets of lactating cows [2]. Therefore, researches paid more attentions on the genetic improvement for milk FA traits in dairy cattle, and studies showed that the heritability of SFA and UFA were 0.14–0.33 and 0.08–0.29, respectively, in Holstein cows [3,4,5,6,7].
There is a growing number of genome-wide association studies (GWASs) on milk FA traits, and many candidate genes were identified [8,9,10,11,12]. Our previous GWAS [12] showed that four significant single nucleotide polymorphisms (SNPs), BTB-01423653, BTB-01423676, Hampmap30570-BTA-152778 and ARS-BFGL-NGS-17676, with the distance of 55 ~ 495 kb from PRLR (Prolactin receptor) were associated with C18index (\( \frac{\mathbf{C18}:\mathbf{1}}{\mathbf{C18}:\mathbf{1}+\mathbf{C}\mathbf{1}\mathbf{8}:\mathbf{0}}\times \mathbf{1}\mathbf{0}\mathbf{0} \)), SFA, UFA and SFA/UFA (P = 2.45E-05 ~ 2.55E-06); ARS-BFGL-NGS-13938, 19.3 kb far away from MOGAT1 (Monoacylglycerol O-acyltransferase 1), was significantly associated with UFA (P = 2.25E-05); BTA-111275-no-rs within the MINPP1 (Multiple inositol-polyphosphate phosphatase 1) was significantly associated with C12:0 (P = 2.39E-05); and Hapmap46411-BTA-15820 within the CHUK (Conserved helix-loop-helix ubiquitous kinase) had strong associations with C14:1 (P = 8.29E-06) and C14index (\( \frac{\mathbf{C14}:\mathbf{1}}{\mathbf{C14}:\mathbf{1}+\mathbf{C}\mathbf{1}\mathbf{4}:\mathbf{0}}\times \mathbf{1}\mathbf{0}\mathbf{0} \) ; P = 1.10E-08). Thus, the four genes were considered as the promising candidates for milk FA traits in Chinese Holstein.
PRLR during the pregnancy may be associated with mammary development [13], and it can guide and maintain mammary epithelial cells for continuous lactation during milking of dairy cows [14]. MOGAT1catalyzes the conversion of monoacylglycerols to diacylglycerols, the precursor of several physiologically important lipids such as phosphatidylethanolamine and triacylglycerol [15]. MOGAT1 expression is inversely correlated with lipolytic rates, and its suppression increases basal lipolytic activity [16]. MINPP1 is a stress protein and it can interact with the glucose-1-phosphate to induce apoptosis [17, 18] and human MINPP1 plays a role in differentiation and apoptosis [19]. CHUK gene is also named IKKA gene, and IKKα is normally an activator of the transcription factor nuclear factor-κB, and it leads to potent activation of SREBP-mediated lipogenesis in the context of hepatitis C virus infection [20].
In this study, we further explored the polymorphisms and genetic effects of PRLR, MOGAT1, MINPP1 and CHUK genes on milk FA traits, and searched the potential functional mutations.
Results
SNPs identification
In this study, we identified four, two, two, and three SNPs in PRLR, MOGAT1, MINPP1, and CHUK genes, respectively (Additional file 2: Table S2). For PRLR, two SNPs (g.38948871C > T and g.38949011G > A) were in the 5′ flanking region, and two SNPs (g.39115344G > A and g.39115345 T > C) were in the exon 4. In MOGAT1, both g.111599360A > G and g.111601747 T > A were in the 5′ flanking region. The g.9206582C > T and g.9207070A > G were observed in the 3′ UTR and the intron 5 of MINPP1, respectively. For CHUK, g.21008688G > T was in the 5′ flanking region, and two SNPs (g.20966385C > G and g.20966354C > T) were in the 3′ UTR. Out of these SNPs, g.39115344G > A in PRLR was a missense mutation with the substitution of amino acid from serine to asparagine when the allele mutated from G to A.
Associations between SNPs/haplotype blocks and milk fatty acids
We performed the association analyses between the total 11 SNPs and 23 milk FA traits, and the results were shown in Additional file 3: Table S3. For PRLR gene, g.38948871C > T was significantly associated with C6:0 (P = 0.0027) and UFA (P = 0.0364); g.38949011G > A was significantly associated with C8:0 (P = 0.0108); g.39115344G > A was significantly associated with C6:0, C8:0, C14:0, C17:1, C17index, SFA, and UFA (P = 0.022 ~ < 0.0001); and g.39115345 T > C was significantly associated with C6:0, C8:0, C10:0, C14:0, C18index, C17index, SFA, and UFA (P = 0.0456 ~ < 0.0001). For MOGAT1 gene, the two SNPs (g.111599360A > G and g.111601747 T > A) were significantly associated with C8:0 (P = 0.0001 and P < 0.0001), and g.111599360A > G was also significantly associated with C18:0 (P = 0.0058) and C17index (P = 0.0153). Both g.9206582C > T and g.9207070A > G of MINPP1 gene had significant associations with C6:0, C8:0, C10:0 and C17:0 (P = 0.0271 ~ < 0.0001), and g.9206582C > T was also significantly associated with C20:0 (P = 0.0436). The association analyses results of three SNPs (g.21008688G > T, g.20966385C > G and g.20966354C > T) of the CHUK showed that they were all significantly associated with C8:0, C10:0, C12:0, C14:0, C16:1, C17:0, C17:1, C18:0, C16index, and C17index (P = 0.0423 ~ < 0.0001). In addition, g.21008688G > T was strongly associated with C18index, C20:0, and UFA (P = 0.0099 ~ 0.0014); g.20966385C > G was strongly associated with C14:1 (P = 0.0368) and C14index (P < 0.0001); and g.20966354C > T had significant associations with C6:0, C18:1cis-9, C18index, C20:0, SFA, UFA, and SFA/UFA (P = 0.0114 ~ < 0.0001). Correspondingly, the significances of additive (а), dominant (d) and substitution (α) effects for the 11 SNPs with milk FA traits were shown in Additional file 4: Table S4.
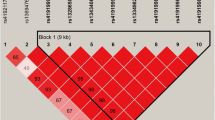
We found five haplotype blocks (D′ = 0.97 ~ 1.00; Fig. 1) in the study, including two in PRLR, one in MOGAT1, one in MINPP1, and one in CHUK. By haplotype-based association analyses (Additional file 5: Table S5), we found that the block 1 in PRLR was significantly associated with C6:0 (P = 0.0389); block 2 in PRLR was significantly associated with C6:0, C14:0, C17:1, C17index, SFA and UFA (P = 0.0392 ~ 0.0001); block 3 in MOGAT1 had significant associations with C8:0, C16:1, C18:0 and C17index (P = 0.0364 ~ < 0.0001); block 4 in MINPP1 had strong associations with C6:0, C8:0, C10:0, C18:1cis-9, C20:0, C17index, and UFA (P = 0.0306 ~ < 0.0001); and block 5 in CHUK was strongly associated with C6:0, C8:0, C10:0, C14:0, C16:1, C17:0, C18:0, C18:1cis-9, C18index, C20:0, C14index, C16index, C17index, SFA, UFA, and SFA/UFA (P = 0.0256 ~ < 0.0001).

The linkage disequilibrium (LD; D′ =0.97 ~ 1.00) estimated among the SNPs in PRLR, MOGAT1, MINPP1 and CHUK genes. Haplotype block 1 included g.38948871C > T and g.38949011G > A in PRLR; haplotype block 2 included g.39115344G > A and g.39115345 T > C in PRLR; haplotype block 3 included g.111599360A > G and g.111601747 T > A in MOGAT1; haplotype block 4 included g.9206582C > T and g.9207070A > G in MINPP1; and haplotype block 5 included g.20966385C > G and g.20966354C > T in CHUK. D′ is the value of D prime between the two loci
Functional variation caused by SNPs of PRLR, MOGAT1, MINPP1 and CHUK
We used the Genomatix software to predicted the changes of TFBSs for all the five SNPs in 5′ flanking region of the four genes, and found that totally three SNPs (g.38949011G > A, g.111599360A > G and g.111601747 T > A) could alter the TFBSs (Fig. 2). The allele G of g.38949011G > A created a BS for the transcription factor MYBL1 (V-myb avian myeloblastosis viral oncogene homolog-like 1; MST = 0.8), and the allele A created the BSs for MEL1 (MDS1/EVI1-like gene 1; MST = 1.0) and LTSM (LTSM elements with 8 bp spacer; MST = 0.8). Alleles A and G of g.111599360A > G created the BSs for PAX6 (Pax-6 paired domain binding site; MST = 0.8) and TEAD4 (TEA domain family member 4; MST = 0.9), respectively. The allele A of g.111601747 T > A created three TFBSs, namely, AP4 (Activating enhancer binding protein 4; MST = 1.0), TCF21 (Transcription factor 21; MST = 1.0) and NEUROG (Neurogenin 1 and 3 binding sites; MST = 0.9).

Changes of transcripton factor binding sites caused by the SNPs in 5′ flanking regions. The SNPs in sequences were highlighted in red
We further used the SOPMA to predict the protein secondary structure variation for the missense mutation (g.39115344G > A) in PRLR, and found that the PRLR protein secondary structure altered from serine (AGT) to asparagine (AAT). It showed that the α-helix was from 16.70 to 14.97%, the extended strand from 21.86 to 21.69%, the β-turn from 4.65 to 3.79%, and the random coil from 56.80 to 59.55%.
In addition, we used the position information of all the SNPs in the 3′ UTR to confirm that whether the SNPs altered the BSs for the microRNAs according to the TargestScanHuman database, and found that the allele C of g.20966385C > G in the 3′ UTR of CHUK was located in the seed region for targeting the specific microRNAs, bta-miR-2392, bta-miR-2434/3602 and bta-miR-2395.
Discussion
The PRLR, MOGAT1, MINPP1 and CHUK genes were considered as the promising candidates for milk FA traits in our previous GWAS [12], and their polymorphisms and genetic effects were determinated in this study.
In recent years, the association between the gene polymorphism and milk production traits in dairy cattle has become a hotspot [21,22,23]. In this study, we found that each SNP of the PRLR, MOGAT1, MINPP1, and CHUK genes had significant association with at least one milk FA trait. Haplotypes formed by the SNPs have important implications for identifying associations with complex traits [24]. For the identified SNPs, we estimated their LD, and found that they were highly linked, which might be due to the properties of each SNP, such as the frequency and population history [25]. The haplotype-based association analyses showed that each haplotype block was also significantly associated with at least one milk FA trait.
From the KEGG database (https://www.genome.jp/kegg/pathway.html), we found that PRLR gene is involved in PI3K-AKT (ko04151) and Jak-STAT (ko04630) signaling pathways, which were identified to be associated with lipid metabolism [26, 27]. In this study, g.38949011G > A in the 5′ flanking region of PRLR altered the TFBSs, in which, the allele G created a BS for and the MYBL1, and the allele A created the BSs for MEL1 and LTSM. As we know, transcription factors (TFs) are the sequence-specific DNA-binding proteins that can regulate gene transcription, and genomic locations at which TFs interact with DNA are considered as TFBSs [28]. Some studies showed that TFs can play the important roles in gene expression [29, 30]. MYBL1 as a DNA-binding TF can bind the mim-1 promoter and to activate its expression to regulate the oncogenesis [31], and MEL1 can stabilize the inactive Smad3-SKI complex on the promoter of TGF-β target genes to inhibit TGF-β signaling [32]. LTSM can enhance the transcriptional activity of the promoters in dependency of the distance from the transcription start site [33]. Hence, we deduced that these TFs might regulate PRLR gene expression to impact the components of milk FAs. In addition, we found a missense mutation in PRLR gene, g.39115344G > A, altered the protein secondary structure. Proteins are the utmost multi-purpose macromolecules (i.e., main catalysts, structural elements, signaling messengers and molecular machines of biological tissues) [34], which play a crucial function in many aspects of biological processes [35]. The protein structure plays a decisive role in function of protein, for example, a study reports that the β-turn is essential for the structure and function of proteins [36]. The prediction of protein structure from amino acid sequences is one of the most vital problems in molecular biology, and the fundamental elements of the protein secondary structure are α-helix, extended strand, β-turn, and coils [37]. In this study, α-helix, extended strand, β-turn, and random coil were all changed because of the mutation from G to A in g.39115344G > A, indicating that this missense mutation might affect the function of PRLR through changing the protein secondary structure.
MOGAT1 gene is involved in the glycerolipid metabolism (ko00561), and codes the MGAT (monoacylglycerol acyltransferases) enzyme, which is active in human liver and its activity can represent a viable target for pharmacologic intervention to treat nonalcoholic fatty liver disease [38]. A study reported that up-regulation of MOGAT1 gene can mediate hepatic steatosis by increasing intracellular diacylglycerol content [39]. In this study, g.111599360A > G in 5′ flanking region of MOGAT1 was predicted to create the TFBSs for PAX6 with the allele A and TEAD4 with the allele G. PAX6 can respectively down-regulate Sox3 and up-regulate Lhx9 in the Pax6-mutant cortex to exert its effects at the molecular level during murine forebrain neurogenesis [40]. TEAD4 can directly induce Myogenin, CDKN1A and Cavelin3 expression to promote myoblast differentiation [41]. Mutation of TAED4 at either site can decrease its occupancy on the promoter region of target genes, and largely impair the target gene transcription to inhibit the growth and colony formation of gastric cancer cell HGC-27 [42]. By association analyses, the cows with AA genotype of g.111599360A > G had higher C8:0 than that with GG, implicating that PAX6 might enhance the content C8:0 by regulating the target gene MOGAT1. While, the TEAD4 might regulate MOGAT1 to reduce C8:0 by binding the G allele. In addition, g.111601747 T > A in the 5′ flanking region of MOGAT1, created the BSs for three TFs, namely, AP4, TCF21, and NEUROG. It is reported that AP4 can up-regulate the expression of LAPTM4B to promote cell growth, migration, invasion, and cisplatin resistance in breast cancer [43]. AP4-geminin complex suppresses the precocious expression of target genes in fetal brain [44]. TCF21 plays a repression role for its target gene to affect the phenotypes [45, 46]. Over-expression of NEUROG can override the pluripotency-specific gene network and force human embryonic stem cells to differentiate into neurons [47]. The cows with AA genotype of g.111601747 T > A had significantly lower C8:0, suggesting that the three TFs (AP4, TCF21 and NEUROG) might work together to regulate the expression of MOGAT1 gene to finally decrease the C8:0.
MINPP1 is involved in the glycolysis/gluconeogenesis (ko00010) and inositol phosphate metabolism (ko00562) signaling pathways. Glycolysis can completely bypass 3-phosphoglycerate through that MINPP1 converts 2,3-bisphosphoglycerate to 2-phosphoglycerate, which activates the AMPK cascade [48]. Furthermore, AMPK can stimulate the fatty acid oxidation [49]. In this study, we identified g.9207070A > G in the intron 5 of the MINPP1 gene. In 1987, the first finding that introns can increase the gene expression was found in maize [50]. Introns include the regulatory regions, that can conferred developmental and cell-specific expression of a gene reside [51]. The rs734553 located on the intron 7 of SLC2A9 in human is greatly related with serum uric acid of healthy individuals with normal renal function, thus it is powerful for prediction of chronic kidney disease progression [52]. Our association analyses showed that the cows with GG genotype of g.9207070A > G had significantly higher C6:0, and lower C17:0. We herein concluded that the intron mutation might be able to affect the milk FA traits that deserved the further validation.
CHUK gene is involved in the MAPK (ko04010), mTOR (ko04150), PI3K-Akt (ko04151), and Adipocytokine (ko04920) signaling pathways associated with lipid metabolism. In 2012, the associations between a SNP (rs11597086) of the human CHUK and lipid phenotype were identified [53]. In this study, the allele C of g.20966385C > G in the 3′ UTR of CHUK is in the seed sequences for targeting the microRNAs (bta-miR-2392, bta-miR-2434/3602 and bta-miR-2395). MicroRNA is a class of gene expression regulating factors and plays an important role in maintaining genome stability, regulating growth and development, and other biological processes [54, 55]. Some studies reported that microRNA regulates the fat metabolism and lipid metabolism disorders through the targeting genes, for example, miR-196 may be related to the gene expression of the homologous genes in subcutaneous adipose tissue and lipid distribution [56]. For the three microRNAs, bta-miR-2392, bta-miR-2434/3602 and bta-miR-2395, there have not been studied to reveal the regulatory function in cattle until now. In human, the miR-2392 can regulate its target gene MAML3 and WHSC1 to suppress metastasis and epithelial-mesenchymal transition in gastric cancer [57]. Our association analyses showed that the cows with CC genotype of g.20966385C > G had significantly higher C14:1, C17:0, C18:0 and C14inex, and lower C14:0, C16:1, C16index and C17index, indicating that the three microRNAs might regulate the expression of CHUK to affect the milk FA traits.
In dairy cattle breeding, the integration of DNA marker technology and genomics into the traditional evaluation system decreased generation interval and increased the selection accuracy, so that the cost of progeny testing was reduced [58]. Here, we found significant SNPs for milk FAs in dairy cattle, which could be used as the genetic markers for the genomic selection to improve the accuracy of selection and lower the breeding cost. This study provided the evidence for associations of PRLR, MOGAT1, MINPP1 and CHUK genes with milk FAs, and the in-depth study should be performed to verify the regulatory mechanism of these genes for milk FAs in dairy cattle by biotechnologies, such as RNA interference and gene editing.
Conclusions
Our findings first confirmed the genetic effects of PRLR, MOGAT1, MINPP1 and CHUK genes on milk FAs in Chinese Holstein. Three SNPs, g.38949011G > A of PRLR, g.111599360A > G and g.111601747 T > A of MOGAT1, might be the functional mutations by changing the promoter activities. In addition, the missense mutation in PRLR, g.39115344G > A, changed the protein secondary structure was suggested to be a critical mutation to affect the PRLR protein function. Furthermore, g.20966385C > G in 3′ UTR of CHUK varied the binding sequences for microRNAs that could regulate the gene expression of CHUK. This study provided the basis for further investigation on function validation of PRLR, MOGAT1, MINPP1 and CHUK genes, and the potential functional mutations might serve as genetic markers to apply to the breeding program for milk FA traits in Chinese Holstein.
Methods
Animals and phenotypic data
A total of 1065 Chinese Holstein cows belonging to 44 sire families were collected from 23 dairy farms of Beijing Sanyuanlvhe Dairy Farming Center (Beijing, China), where the standard performance testing for dairy herd improvement (DHI) has been regularly conducted since 1999. These cows were full blood of Chinese Holstein breed that were originated from cross-breeding between the Chinese Yellow cattle and European Holstein, over the past 100 years, by continuous import of foreign Holstein bulls, semen and embryos, mainly from USA and a few from Canada and Europe, which were directly used to AI or used to cross Chinese Holstein cows through planned mating to generate breeding bulls [59]. All the cows were fed with the same regular total mixed ration composed of concentrated feed and coarse fodder. We obtained 50 ml milk samples during November to December of 2014 to measure the DHI, and then the remaining milk were frozen in the − 20 °C to be used for detecting the milk FAs in the laboratory of the Beijing Dairy Cattle Center (www.bdcc.com.cn). We used gas chromatography to measure the 16 FAs (C6:0, C8:0, C10:0, C11:0, C12:0, C13:0, C14:0, C15:0, C16:0, C17:0, C18:0, C20:0, C14:1, C16:1, C17:1, and C18:1cis-9) in the milk samples according to the descriptions of procedures in the previous studies [12, 60], and calculated C14index, C16index, C17index and C18index using the formula [61]: \( \frac{\mathrm{cis}-9\ \mathrm{unsaturated}}{\mathrm{cis}-9\ \mathrm{unsaturated}+\mathrm{saturated}}\times 100 \). In addition, we summarized SFA, UFA and SFA/UFA.
DNA extraction and SNP identification
We used the TIANamp Blood DNA kit (Tiangen, Beijing, China) and salt-out procedure to extract the genomic DNA form the blood samples of 1065 Chinese Holsteins and the semen samples of 44 sires, respectively. Then the quantity and quality of the genomic DNAs were measured by NanoDrop 2000 Spectrophotometer (Thermo Scientific, DE, USA) and gel electrophoresis, respectively.
We designed a total of 85 primers (Additional file 1: Table S1) in the entire exons with their partial adjacent introns, and 3000 bp of 5′ and 3′ flanking regions of the PRLR, MOGAT1, MINPP1, and CHUK genes using the Primer 3 (http://bioinfo.ut.ee/primer3-0.4.0/) according to their bovine genomic sequences (GenBank accession no. AC_000177.1, AC_000159.1, AC_000183.1 and AC_000183.1). The primers were synthesized in Beijing Genomics Institute (BGI, Beijing, China). The 44 semen DNAs were randomly divided into two DNA pools (22 DNA in each pool) for the PCR amplification, and the concentration of each DNA was 50 ng/μL. PCR conditions were as follows: initial denaturation at 94 °C for 5 min, followed by 35 cycles at 94 °C for 30s; 60 °C for 30s; 72 °C for 30s; and a final extension at 72 °C for 7 min. Afterwards, we sequenced the PCR products using ABI3730XL DNA analyzer (Applied Biosystems, Foster, CA, USA), and aligned the sequences with the bovine reference sequence (UMD 3.1) by the BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) for identifying the potential SNPs.
Genotyping and linkage disequilibrium (LD) analyses
Genotypes of the identified SNPs were obtained from 1065 cows with Sequenom MassArray by matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS, Sequenom MassARAY, Bioyong Technologies Inc., HK). In addition, we used the Haploview 4.1 (Broad Insititute, Cambridge, MA, USA) to analyze the LD extent among the identified SNPs.
Association analyses
The association analyses between each SNP/haplotype block and 23 milk FA traits were conducted with SAS9.2 (SAS Institute Inc., Cary, NC, USA) using the following model:
For each trait, Yijklm was the phenotypic value of the 1065 cows; μ was the overall mean; Gi, hj lk and Mm were the fixed effects of the genotypes or haplotypes, farm (23), stage of lactation (4) and calving months (293), respectively; al was the random polygenic effect; b represented the regression coefficient of covariate M; and eijklm was the random residual. We considered a significant association when the P was less than 0.05/N, where N was the number of genotypes or haplotype combinations. In addition, we calculated the additive effect (a), dominant effect (d), and substitution effect (α) according to the formulas by Falconer & Mackay [62]: a = (AA − BB)/2, d = AB − (AA + BB)/2 and a = a + d(q − p), where AA, AB and BB represent the square means of milk FA traits corresponding to the genotypes, p and q refer to the allele frequencies of corresponding loci.
Biological function prediction
We used the Genomatix software (http://www.genomatix.de/cgi-bin/welcome/welcome.pl?s=d1b5c9a9015b02bb3b1a806f9c03293f) to predict whether the SNPs in the 5′ flanking region of the PRLR, MOGAT1, MINPP1 and CHUK genes altered the transcription factor binding sites (TFBSs; matrix similarity threshold, MST > 0.8). Furthermore, we explored the changes of protein secondary structure for missense mutation by the SOPMA software (https://prabi.ibcp.fr/htm/site/web/services/secondaryStructurePrediction#SOPMA). For the SNPs in the 3′ untranslated region (UTR), we aligned them to the TargetScanHuman database (http://www.targetscan.org/vert_71/) for researching whether they changed the binding sites (BSs) of seed sequences with the microRNAs.
Availability of data and materials
All relevant data are available within the article and its additional files.
Abbreviations
- a:
-
Additive effect
- BS:
-
Binding site
- CHUK:
-
Conserved helix-loop-helix ubiquitous kinase
- d:
-
Dominant effect
- DHI:
-
Dairy herd improvement
- FA:
-
Fatty acids
- GWAS:
-
Genome-wide association study
- LD:
-
Linkage disequilibrium
- MINPP1:
-
Multiple inositol-polyphosphate phosphatase 1
- MOGAT1:
-
Monoacylglycerol O-acyltransferase 1
- MST:
-
Matrix similarity threshold
- PRLR:
-
Prolactin receptor
- SFA:
-
Saturated fatty acids
- SNP:
-
Single nucleotide polymorphism
- TF:
-
Transcription factor
- TFBS:
-
Transcription factor binding site
- UFA:
-
Unsaturated fatty acids
- UTR:
-
Untranslated region
- α:
-
Substitution effect
References
Melfsen A, Holstermann M, Haeussermann A, Molkentin J, Susenbeth A, Hartung E. Accuracy and application of milk fatty acid estimation with diffuse reflectance near-infrared spectroscopy. J Dairy Res. 2018;85(2):212–21.
Grummer RR. Effect of feed on the composition of milk fat. J Dairy Sci. 1991;74(9):3244–57.
Krag K, Poulsen NA, Larsen MK, Larsen LB, Janss LL, Buitenhuis B. Genetic parameters for milk fatty acids in Danish Holstein cattle based on SNP markers using a Bayesian approach. BMC Genet. 2013;14:79.
Petrini J, Iung LH, Rodriguez MA, Salvian M, Pertille F, Rovadoscki GA, Cassoli LD, Coutinho LL, Machado PF, Wiggans GR, et al. Genetic parameters for milk fatty acids, milk yield and quality traits of a Holstein cattle population reared under tropical conditions. J Anim Breed Genet = Z Tierzuecht Zuechtungsbiol. 2016;133(5):384–95.
Narayana SG, Schenkel FS, Fleming A, Koeck A, Malchiodi F, Jamrozik J, Johnston J, Sargolzaei M, Miglior F. Genetic analysis of groups of mid-infrared predicted fatty acids in milk. J Dairy Sci. 2017;100(6):4731–44.
Stoop WM, Schennink A, Visker MH, Mullaart E, van Arendonk JA, Bovenhuis H. Genome-wide scan for bovine milk-fat composition. I. Quantitative trait loci for short- and medium-chain fatty acids. J Dairy Sci. 2009;92(9):4664–75.
Schennink A, Stoop WM, Visker MH, van der Poel JJ, Bovenhuis H, van Arendonk JA. Short communication: genome-wide scan for bovine milk-fat composition. II. Quantitative trait loci for long-chain fatty acids. J Dairy Sci. 2009;92(9):4676–82.
Palombo V, Milanesi M, Sgorlon S, Capomaccio S, Mele M, Nicolazzi E, Ajmone-Marsan P, Pilla F, Stefanon B, D'Andrea M. Genome-wide association study of milk fatty acid composition in Italian Simmental and Italian Holstein cows using single nucleotide polymorphism arrays. J Dairy Sci. 2018.
Buitenhuis B, Janss LL, Poulsen NA, Larsen LB, Larsen MK, Sorensen P. Genome-wide association and biological pathway analysis for milk-fat composition in Danish Holstein and Danish Jersey cattle. BMC Genomics. 2014;15:1112.
Li X, Buitenhuis AJ, Lund MS, Li C, Sun D, Zhang Q, Poulsen NA, Su G. Joint genome-wide association study for milk fatty acid traits in Chinese and Danish Holstein populations. J Dairy Sci. 2015;98(11):8152–63.
Knutsen TM, Olsen HG, Tafintseva V, Svendsen M, Kohler A, Kent MP, Lien S. Unravelling genetic variation underlying de novo-synthesis of bovine milk fatty acids. Sci Rep. 2018;8(1):2179.
Li C, Sun DX, Zhang SL, Wang S, Wu XP, Zhang Q, Liu L, Li YH, Qiao L. Genome Wide Association Study Identifies 20 Novel Promising Genes Associated with Milk Fatty Acid Traits in Chinese Holstein. PLoS One. 2014;9(5).
Zi XD, Chen DW, Wang HM. Molecular characterization, mRNA expression of prolactin receptor (PRLR) gene during pregnancy, nonpregnancy in the yak (Bos grunniens). Gen Comp Endocrinol. 2012;175(3):384–8.
Hennighausen L, Robinson GW. Information networks in the mammary gland. Nat Rev Mol Cell Biol. 2005;6(9):715–25.
Sankella S, Garg A, Agarwal AK. Characterization of the Mouse and Human Monoacylglycerol O-Acyltransferase 1 (Mogat1) Promoter in Human Kidney Proximal Tubule and Rat Liver Cells. PLoS One. 2016;11(9).
Liss KHH, Lutkewitte AJ, Pietka T, Finck BN, Franczyk M, Yoshino J, Klein S, Hall AM. Metabolic importance of adipose tissue monoacylglycerol acyltransferase 1 in mice and humans. J Lipid Res. 2018;59(9):1630–9.
Kilaparty SP, Agarwal R, Singh P, Kannan K, Ali N. Endoplasmic reticulum stress-induced apoptosis accompanies enhanced expression of multiple inositol polyphosphate phosphatase 1 (Minpp1): a possible role for Minpp1 in cellular stress response. Cell Stress Chaperones. 2016;21(4):593–608.
Kilaparty SP, Singh A, Baltosser WH, Ali N. Computational analysis reveals a successive adaptation of multiple inositol polyphosphate phosphatase 1 in higher organisms through evolution. Evol Bioinforma. 2014;10:239–50.
Dahia PM, Gimm O, Chi HB, Marsh DJ, Reynolds PR, Eng C. Absence of germline mutations in MINPP1, a phosphatase encoding gene centromeric of PTEN, in patients with Cowden and Bannayan-Riley-Ruvalcaba syndrome without germline PTEN mutations. J Med Genet. 2000;37(9):715–7.
Camus G, Ott M. How the antiviral immune response boosts liver fat. Nat Med. 2013;19(6):671–2.
Han B, Liang W, Liu L, Li Y, Sun D. Determination of genetic effects of ATF3 and CDKN1A genes on milk yield and compositions in Chinese Holstein population. BMC Genet. 2017;18(1):47.
Viale E, Tiezzi F, Maretto F, De Marchi M, Penasa M, Cassandro M. Association of candidate gene polymorphisms with milk technological traits, yield, composition, and somatic cell score in Italian Holstein-Friesian sires. J Dairy Sci. 2017;100(9):7271–81.
Han B, Liang W, Liu L, Li Y, Sun D. Genetic association of the ACACB gene with milk yield and composition traits in dairy cattle. Anim Genet. 2018;49(3):169–77.
Patil N, Berno AJ, Hinds DA, Barrett WA, Doshi JM, Hacker CR, Kautzer CR, Lee DH, Marjoribanks C, McDonough DP, et al. Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science. 2001;294(5547):1719–23.
Daly MJ, Rioux JD, Schaffner SF, Hudson TJ, Lander ES. High-resolution haplotype structure in the human genome. Nat Genet. 2001;29(2):229–32.
Tawfik MK, El-Kherbetawy MK, Makary S. Cardioprotective and anti-Aggregatory effects of Levosimendan on isoproterenol-induced myocardial injury in high-fat-fed rats involves modulation of PI3K/Akt/mTOR signaling pathway and inhibition of apoptosis: comparison to Cilostazol. J Cardiovasc Pharmacol T. 2018;23(5):456–71.
Gurzov EN, Stanley WJ, Pappas EG, Thomas HE, Gough DJ. The JAK/STAT pathway in obesity and diabetes. FEBS J. 2016;283(16):3002–15.
Fornes O, Gheorghe M, Richmond PA, Arenillas DJ, Wasserman WW, Mathelier A. MANTA2, update of the mongo database for the analysis of transcription factor binding site alterations. Sci data. 2018;5:180141.
Xu ZH, Yoshida T, Wu LJ, Maiti D, Cebotaru L, Duh EJ. Transcription factor MEF2C suppresses endothelial cell inflammation via regulation of NF-kappa B and KLF2. J Cell Physiol. 2015;230(6):1310–20.
Zhang M, Jin X, Chen Y, Wei M, Liao W, Zhao S, Fu C, Yu L. TcMYC2a, a basic helix-loop-helix transcription factor, Transduces JA-Signals and Regulates Taxol Biosynthesis in Taxus chinensis. Front Plant Sci. 2018;9:863.
George OL, Ness SA. Situational awareness: regulation of the myb transcription factor in differentiation, the cell cycle and oncogenesis. Cancers. 2014;6(4):2049–71.
Takahata M, Inoue Y, Tsuda H, Imoto I, Koinuma D, Hayashi M, Ichikura T, Yamori T, Nagasaki K, Yoshida M, et al. SKI and MEL1 cooperate to inhibit transforming growth factor-beta signal in gastric cancer cells. J Biol Chem. 2009;284(5):3334–44.
Roepcke S, Stahlberg S, Klein H, Schulz MH, Theobald L, Gohlke S, Vingron M, Walther DJ. A tandem sequence motif acts as a distance-dependent enhancer in a set of genes involved in translation by binding the proteins NonO and SFPQ. BMC Genomics. 2011;12:624.
Eisenberg D, Marcotte EM, Xenarios I, Yeates TO. Protein function in the post-genomic era. Nature. 2000;405(6788):823–6.
Panda B, Majhi B, Thakur A. An integrated-OFFT model for the prediction of protein secondary structure. Curr Comput Aided Drug Des. 2018.
Mensch C, Johannessen C. Is Raman optical activity spectroscopy sensitive to beta-turns in proteins? Secondary structure and side-chain dependence. Chemphyschem. 2018.
Kouza M, Faraggi E, Kolinski A, Kloczkowski A. The GOR method of protein secondary structure prediction and its application as a protein aggregation prediction tool. Methods Mol Biol. 2017;1484:7–24.
Hall AM, Kou K, Chen ZJ, Pietka TA, Kumar M, Korenblat KM, Lee K, Ahn K, Fabbrini E, Klein S, et al. Evidence for regulated monoacylglycerol acyltransferase expression and activity in human liver. J Lipid Res. 2012;53(5):990–9.
Ramon-Krauel M, Pentinat T, Bloks VW, Cebria J, Ribo S, Perez-Wienese R, Vila M, Palacios-Marin I, Fernandez-Perez A, Vallejo M, et al. Epigenetic programming at the Mogat1 locus may link neonatal overnutrition with long-term hepatic steatosis and insulin resistance. FASEB J. 2018. https://doi.org/10.1096/fj.201700717RR.
Holm PC, Mader MT, Haubst N, Wizenmann A, Sigvardsson M, Gotz M. Loss- and gain-of-function analyses reveal targets of Pax6 in the developing mouse telencephalon. Mol Cell Neurosci. 2007;34(1):99–119.
Benhaddou A, Keime C, Ye T, Morlon A, Michel I, Jost B, Mengus G, Davidson I. Transcription factor TEAD4 regulates expression of myogenin and the unfolded protein response genes during C2C12 cell differentiation. Cell Death Differ. 2012;19(2):220–31.
Shi Z, He F, Chen M, Hua L, Wang W, Jiao S, Zhou Z. DNA-binding mechanism of the hippo pathway transcription factor TEAD4. Oncogene. 2017;36(30):4362–9.
Wang L, Meng Y, Xu JJ, Zhang QY. The transcription factor AP4 promotes oncogenic phenotypes and cisplatin resistance by regulating LAPTM4B expression. Mol Cancer Res. 2018;16(5):857–68.
Kim MY, Jeong BC, Lee JH, Kee HJ, Kook H, Kim NS, Kim YH, Kim JK, Ahn KY, Kim KK. A repressor complex, AP4 transcription factor and geminin, negatively regulates expression of target genes in nonneuronal cells. Proc Natl Acad Sci U S A. 2006;103(35):13074–9.
Franca MM, Ferraz-de-Souza B, Lerario AM, Fragoso MC, Lotfi CF. POD-1/TCF21 reduces SHP expression, affecting LRH-1 regulation and cell cycle balance in adrenocortical and Hepatocarcinoma tumor cells. Biomed Res Int. 2015;2015:841784.
Chen B, Zeng C, Ye Y, Wu D, Mu Z, Liu J, Xie Y, Wu H. Promoter methylation of TCF21 may repress autophagy in the progression of lung cancer. J Cell Commun Signal. 2018;12(2):423–32.
Matsushita M, Nakatake Y, Arai I, Ibata K, Kohda K, Goparaju SK, Murakami M, Sakota M, Chikazawa-Nohtomi N, Ko SBH, et al. Neural differentiation of human embryonic stem cells induced by the transgene-mediated overexpression of single transcription factors. Biochem Biophys Res Commun. 2017;490(2):296–301.
Cho J, King JS, Qian X, Harwood AJ, Shears SB. Dephosphorylation of 2,3-bisphosphoglycerate by MIPP expands the regulatory capacity of the Rapoport-Luebering glycolytic shunt. Proc Natl Acad Sci U S A. 2008;105(16):5998–6003.
Ballester M, Amills M, Gonzalez-Rodriguez O, Cardoso TF, Pascual M, Gonzalez-Prendes R, Panella-Riera N, Diaz I, Tibau J, Quintanilla R. Role of AMPK signalling pathway during compensatory growth in pigs. BMC Genomics. 2018;19(1):682.
Callis J, Fromm M, Walbot V. Introns increase gene-expression in cultured maize cells. Genes Dev. 1987;1(10):1183–200.
Reddy VS, Reddy ASN. Developmental and cell-specific expression of ZWICHEL is regulated by the intron and exon sequences of its upstream protein-coding gene. Plant Mol Biol. 2004;54(2):273–93.
Yi XL, Li J, Meng DM, Liu YJ, Liu YH, Ma HM, Yuan Y, Xing SC. An intron variant of SLC2A9 increases the risk for type 2 diabetes mellitus complicated with hyperuricemia in Chinese male population. Iran J Public Health. 2018;47(6):844–51.
Asselbergs FW, Guo Y, van Iperen EP, Sivapalaratnam S, Tragante V, Lanktree MB, Lange LA, Almoguera B, Appelman YE, Barnard J, et al. Large-scale gene-centric meta-analysis across 32 studies identifies multiple lipid loci. Am J Hum Genet. 2012;91(5):823–38.
Ruiz-Ferrer V, Voinnet O. Roles of plant small RNAs in biotic stress responses. Annu Rev Plant Biol. 2009;60:485–510.
Bonnet E, Van de Peer Y, Rouze P. The small RNA world of plants. New Phytol. 2006;171(3):451–68.
Divoux A, Xie H, Li JL, Karastergiou K, Perera RJ, Chang RJ, Fried SK, Smith SR. MicroRNA-196 regulates HOX gene expression in human gluteal adipose tissue. Obesity. 2017;25(8):1375–83.
Li JJ, Li TY, Lu YY, Shen GF, Guo H, Wu J, Lei C, Du F, Zhou FL, Zhao XD, et al. MiR-2392 suppresses metastasis and epithelial- mesenchymal transition by targeting MAML3 and WHSC1 in gastric cancer. FASEB J. 2017;31(9):3774–86.
Wiggans GR, Cole JB, Hubbard SM, Sonstegard TS. Genomic selection in dairy cattle: the USDA experience. Annu Rev Anim Biosci. 2017;5:309–27.
Sun D, Jia J, Ma Y, Zhang Y, Wang Y, Yu Y, Zhang Y. Effects of DGAT1 and GHR on milk yield and milk composition in the Chinese dairy population. Anim Genet. 2009;40(6):997–1000.
Shi L, Han B, Liu L, Lv X, Ma Z, Li C, Xu L, Li Y, Zhao F, Yang Y, et al. Determination of Genetic Effects of LIPK and LIPJ Genes on Milk Fatty Acids in Dairy Cattle. Genes. 2019;10(2).
Kelsey JA, Corl BA, Collier RJ, Bauman DE. The effect of breed, parity, and stage of lactation on conjugated linoleic acid (CLA) in milk fat from dairy cows. J Dairy Sci. 2003;86(8):2588–97.
Falconer DS, Mackay TFC: Introduction to quantitative genetics. 1996: Ed. 4 xv + 464 pp.
Acknowledgements
We appreciate Beijing Dairy Cattle Center, Beijing Municipal Bureau of Agriculture, and Beijing Sanyuanlvhe Dairy Farming Center, for providing the milk, semen, and blood samples of Chinese Holstein.
Funding
This work was financially supported by the National Natural Science Foundation of China (31802041, 31872330, 31472065, and 31072016), Beijing Dairy Industry Innovation Team (BAIC06–2017/2018), National Science and Technology Programs of China (2013AA102504 and 2014ZX08009-053B), Beijing Natural Science Foundation (6152013), and the Program for Changjiang Scholar and Innovation Research Team in University (IRT_15R62).
Author information
Authors and Affiliations
Contributions
BH and DS conceived and designed the experiments, LL, ZM and YY prepared the milk, blood and semen samples, LS extracted the DNA for SNP identification and genotyping with the help of, XL, YL, and ZF, XL measured the milk fatty acids, LS analyzed the data, and the manuscript was prepared by LS, DS and BH. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All protocols for collection of the tissues of experimental individuals and phenotypic observations were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at China Agricultural University. Tissue samples were collected specifically for this study following standard procedures with the full agreement of the Beijing Sanyuanlvhe Dairy Farming Center who owned the Holstein cows and bulls, respectively.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Details of PCR primers of PRLR, MOGAT1, MINPP1, and CHUK genes. (XLSX 20 kb)
Additional file 2:
Table S2. Information about the 11 identified SNPs. (XLSX 10 kb)
Additional file 3:
Table S3. Associations of 11 SNPs of PRLR, MOGAT1, MINPP1 and CHUK genes with fatty acid traits in Chinese Holstein (LSM ± SE). (XLSX 26 kb)
Additional file 4:
Table S4. Additive (a), dominant (d), and allele substitution (α) effects of 11 SNPs on milk fatty acid traits in Chinese Holstein cows. (XLSX 18 kb)
Additional file 5:
Table S5. Associations of the haplotype blocks with milk fatty acid traits in Chinese Holstein (LSM ± SE). (XLSX 19 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Shi, L., Liu, L., Lv, X. et al. Polymorphisms and genetic effects of PRLR, MOGAT1, MINPP1 and CHUK genes on milk fatty acid traits in Chinese Holstein. BMC Genet 20, 69 (2019). https://doi.org/10.1186/s12863-019-0769-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-019-0769-1