
Abstract
Background
Ketosis in dairy cattle has been shown to cause a high morbidity in the farm and substantial financial losses to dairy farmers. Ketosis symptoms, however, are difficult to identify, therefore, the amount of ketone bodies (mainly β-hydroxybutyric acid, BHB) is used as an indicator of subclinical ketosis in cows. It has also been shown that milk BHB concentrations have a strong correlation with ketosis in dairy cattle. Mid-infrared spectroscopy (MIR) has recently became a fast, cheap and high-throughput method for analyzing milk components. The aim of this study was to perform a genome-wide association study (GWAS) on the MIR-predicted milk BHB to identify genomic regions, genes and pathways potentially affecting subclinical ketosis in North American Holstein dairy cattle.
Results
Several significant regions were identified associated with MIR-predicted milk BHB concentrations (indicator of subclinical ketosis) in the first lactation (SCK1) and second and later lactations (SCK2) in Holstein dairy cows. The strongest association was located on BTA6 for SCK1 and BTA14 on SCK2. Several SNPs on BTA6 were identified in regions and variants reported previously to be associated with susceptibility to ketosis and clinical mastitis in Jersey and Holstein dairy cattle, respectively. One highly significant SNP on BTA14 was found within the DGAT1 gene with known functions on fat metabolism and inflammatory response in dairy cattle. A region on BTA6 and three SNPs on BTA20 were found to overlap between SCK1 and SCK2. However, a novel region on BTA20 (55–63 Mb) for SCK2 was also identified, which was not reported in previous association studies. Enrichment analysis of the list of candidate genes within the identified regions for MIR-predicted milk BHB concentrations yielded molecular functions and biological processes that may be involved in the inflammatory response and lipid metabolism in dairy cattle.
Conclusions
The results of this study confirmed several SNPs and genes identified in previous studies as associated with ketosis susceptibility and immune response, and also found a novel region that can be used for further analysis to identify causal variations and key regulatory genes that affect clinical/ subclinical ketosis.
Similar content being viewed by others
Background
The increase in milk production as a result of intense genetic selection in dairy cattle has been accompanied by a higher incidence of reproductive health issues and production-related diseases, including metritis, ketosis and fatty liver [1, 2]. This can be due to the metabolic changes and challenges in high-producing dairy cows early in lactation and the failure of the cows in maintaining their internal homeostatic and homeorhetic regulations [3]. It has been reported that approximately 50% of the metabolic and infectious diseases occur during the postpartum and transition period in dairy cattle [4, 5]. Reports of eleven studies carried out in several countries have shown that the median incidence of ketosis (per cow and year) was 3.2% for the Holstein breed [6,7,8,9], which can cause a high morbidity in the farm and substantial financial losses to the dairy farmers [10, 11]. Ketosis leads to hypoglycemia and hyperketonemia, and has the highest rate of prevalence after calving that extends to the lactation peak [12, 13]. This metabolic disease can be diagnosed through clinical signs in dairy cattle, including decrease in appetite, weight loss and decrease in milk production [14]. However, these symptoms are usually difficult to detect; therefore, the amount of ketone bodies (acetone, β-hydroxybutyric acid (BHB), and acetoacetate) present in blood, milk, urine and lymph is used as good indicators of subclinical cases in cows [11, 15]. Subclinical ketosis has been defined as when the cow shows a blood BHB level of > 1.2 mmol/L [16, 17] or a non-esterified fatty acid (NEFA) concentration of > 0.4 mM pre-partum or > 1.0 mM postpartum [18]. The gold standard diagnosis of clinical and subclinical ketosis is generally BHB in blood; however, routine blood sampling is not easy to implement, expensive for the farmers [6] and stressful on the cow side [19]. Several studies showed that there may be value in measuring BHB and acetone in milk, which are closely associated with ketosis in cattle [20,21,22]. Mid-infrared spectroscopy (MIR) has recently become a fast, cheap and high-throughput method for analyzing chemicals in livestock and food sectors [23]. The MIR technique uses the absorption of electromagnetic radiation by a sample to determine its chemical composition and has been used to predict milk quality traits since 1960 [24]. It has been shown that the genetic correlation between ketosis and MIR predicted milk BHB in dairy cattle is strong, about 0.75 [8, 25]. Additionally, the prediction accuracy of milk BHB concentrations using fourier transform mid-infrared (FTMIR) was reported to be 71% [26].
Many studies have investigated the metabolic pathways, candidate genes and gene-networks that affect metabolic diseases (including ketosis) or metabolic energy balance in dairy cows pre and post-partum [1, 15, 27, 28]. Additionally, several genes were found to be associated with ketosis biomarkers in dairy cows [15]. However, significant genome-wide regions and genes harboring putative causative mutations have not been reported in previous studies, using the predicted milk BHB concentrations. The objective of this study was 1) to identify genome-wide regions associated with MIR predicted BHB concentrations in milk, as an indicator of sub-clinical ketosis in North American Holstein dairy cattle, and 2) to perform enrichment analysis to identify biologically significant genes and pathways associated with MIR predicted BHB concentrations in milk, as an indicator of subclinical ketosis, and their possible associations with other metabolic correlated traits.
Results
Association analysis
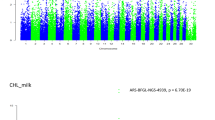
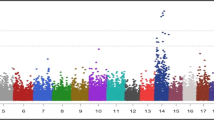
Association analyses using a single SNP regression mixed linear model identified strong associations for the MIR predicted milk BHB from the first (SCK1) and later lactations (SCK2). The –log10 (P-value) of the tested SNPs from the GWAS analyses are shown as Manhattan plots for SCK1 (Fig. 1a) and SCK2 (Fig. 1b). The number of significant SNPs identified at a 5% genome-wise FDR varied from 71 for SCK1 to 369 SNPs for SCK2 (Additional file 1). The Quantile-Quantile (Q-Q) plots and genomic inflation statistics (lambda) of the two GWAS analyses are shown in Fig. 2a and Fig. 2b for SCK1 and SCK2, respectively. The plots showed no evidence of inflation of model-based statistics with a λmedian = 0.986 for SCK1 and λmedian = 0.964 for SCK2 traits.

Genome-wide association analysis using single SNP regression mixed linear model for subclinical ketosis. The log10 of the P-value for association with SNPs is plotted for A. subclinical ketosis in first lactation (SCK1) and B. subclinical ketosis in later lactations (SCK2). Chromosome number is shown on the horizontal axis. The red line is the threshold for significant SNPs at 1% FDR. The green line is the threshold for significant SNPs at 5% FDR

Quantile-quantile (Q-Q) of P-values of SNPs from single SNP regression mixed linear model for subclinical ketosis. The blue dots represent the –log10(P-values) of the observed test statistics for no association for a. subclinical ketosis in first lactation (SCK1) and b. subclinical ketosis in later lactations (SCK2). The red line denotes the expected pattern under the null hypothesis. Deviations between the red line and blue dots indicate how the observed probability of the test statistics deviate from the expected
Annotations and network-based analysis
Annotation of the genes to the corresponding significant SNPs from the GWAS resulted in identification of nine unique genes for SCK1 (Table 1) and 86 unique genes for SCK2 (Table 2). Gene-network analysis was performed using the IPA tool. Using two gene lists, the IPA analysis produced two putative networks for SCK1 and seven networks for the SCK2. The top two putative gene-networks for the SCK1 were associated with disease and functions involved in hematological diseases, organismal injuries and abnormalities, and infectious diseases. For the SCK2, several of the networks resulting from IPA were related to general molecular and cellular processes, including cell death and survival, molecular transport and cellular development. Most of these networks were also involved in more specific pathways such as developmental disorders, embryonic development, endocrine disorders, and metabolic diseases. The most informative networks and molecular interactions, in the context of this study, were the networks, interacting molecules and genes grouped around interferon gamma (IFNG), and tumor necrosis factor (TNF) with IPA score of 20 for SCK1 (Fig. 3), and leptin (LEP) gene with IPA score 12 for SCK2 (Fig. 4).

Network of interactions between GWAS candidate genes for subclinical ketosis in first lactation (SCK1) using ingenuity pathway analysis (IPA). The network shows direct and indirect interactions between the candidate genes enriched for significantly associated SNPs (FDR < 5%) with two gene hubs tumor necrosis factor (TNF) and interferon gamma (IFNG) as two important immune system regulators. The red label indicates the genes that were enriched for significantly associated SNPs. All the relationships were obtained using information contained in the IPA repository

Network of interactions between GWAS candidate genes for subclinical ketosis in later lactations (SCK2) using ingenuity pathway analysis (IPA). The network shows direct and indirect interactions between the candidate genes enriched for significantly associated SNPs (FDR < 5%) with two gene hubs tumor necrosis factor (TNF) and leptin (LEP) as two important genes involved in immune system and lipid metabolism regulators. The red label indicates the genes that were enriched for significantly associated SNPs. All the relationships were obtained using information contained in the IPA repository
The candidate SNP enrichment analysis resulted in enrichment of seven biologically significant GO terms for SCK1 and 91 significant GO terms for SCK2 (Additional file 2), potentially involved in pathways affecting subclinical ketosis. The semantic similarities among the enriched GO terms (FDR < 1%) using REVIGO is shown in Fig. 5.

Gene Ontology scatterplot constructed using REVIGO in R [37] for all the GO terms and genes within the identified significant regions for subclinical ketosis. Settings used for REVIGO program were as follow: database: Bos taurus, semantic similarity: 0.5 (small), semantic similarity measure: SimRel. Colors indicate the P-value of the enriched GO terms provided by the authors. The size of each bubble shows the GO terms with more significant P-values. The size of the bubbles indicates the frequency of the GO terms
Discussion
Subclinical ketosis in first lactation (SCK1)
Most of the identified significant SNPs (FDR < 1%) for SCK1 in this study were located on Bos taurus autosome (BTA) 6 (Fig. 1a, Additional file 1). These SNPs were located at around 88.4~94.8 Mb and this region has not been reported to be significantly associated with ketosis or subclinical ketosis in previous investigations. The long length of regions identified here might be associated with the imputation process. As a result of imputing, missing SNPs and more SNPs and haplotypes (in possible linkage-disequilibrium (LD) with the casual mutations) are added to the dataset. In addition, a Holstein cattle sample commonly has close relatives, who share long haplotypes. Having long haplotypes in LD with the causal mutation might be a reason for the long QTL region identified by the GWAS.
One single significant SNP (FDR < 5%) on BTA6 (BOVINEHD0600015345: rs109930261) was located at 56 Mb (Additional file 1). The position of this SNP is close to the position of several SNPs reported in a recent study on ketosis susceptibility in US Jersey cattle [29]. Parker et al. (2018) reported that the largest proportion of the variance for the ketosis was associated with a region on chromosome 6 at 56.1 Mb [29]. This region was shown to be associated with the genes and functions that affect basal glucose uptake and regulation of glucose transporter GLUT1 and lipid droplet formation [29, 30]. Association of this region on chromosome 6 with milk metabolites, glucose and glutamate, and left-sided displaced abomasum has been also reported in Danish [31] and German [32] Holstein cattle, respectively. Another highly significant (FDR < 1%) SNP identified on BTA6 for SCK1 (BovineHD0600024791: rs109793149) at around 90.5 Mb is located in the gene C-X-C motif chemokine ligand 8 (CXCL8). The protein encoded by this gene is a major mediator of the inflammatory response, secreted primary by neutrophils and acts at the site of infection (Gene ID: 3576). A study of the effect of the ketone body BHB on innate defense capability of the bovine mammary epithelial cells showed that expression of this gene (CXCL8) was significantly induced 30 h post infection with E. coli bacteria [28]. The result of this study is in agreement with previous researches reporting important interactions between metabolic disease and immune response in dairy cow [33, 34]. The association of several genes with key functions in immune response has also been reported in a previous study [29]. Several other candidate genes, including SLC4A4, GC, NPFFR2, ADAMTS3, RASSF6 and MTHFD2L, were identified in this study on BTA6, located within the region 88~90 Mb. The SLC4A4 gene (Solute carrier Family 4, Member 4) is a protein coding gene that encodes a sodium bicarbonate cotransporter (Na+/HCO− 3 cotransporter) involved in the regulation of bicarbonate secretion, absorption and intracellular pH (ID: 8671). The importance of this gene along with other ketogenic genes in various metabolic pathways as well as intracellular pH control in the rumen epithelium tissue was previously reported in neonatal Holstein calves [35]. Another region for milk BHB concentration harbors a functional candidate gene named GC gene. A fine mapping study of a QTL on bovine chromosome 6 using imputed full sequence data suggested that the GC gene has a key role in clinical mastitis and milk production in dairy cattle [36]. The GC gene encodes vitamin D-binding protein (DBP) which is the main carrier of vitamin D3. In both human and bovine, during infection, monocytes and macrophages express and produce and enzyme that converts vitamin D to its biologically active form (25(OH)D3), which has an important role in the immune system and host defense of the animal [36,37,38]. Moreover, concentrations of 25(OH)D3 are reported to be associated with pregnancy cycle and lactation stage [39]. The NPFFR2 gene (neuropeptide FF receptor 2) along with GC and ADAMTS3 (ADAM metallopeptidase with thrombospondin type 1 motif 3) genes were reported previously to be located within a QTL region affecting clinical mastitis in Norwegian Red dairy cattle [36]. Several regions and genes identified in this study associated with BHB in milk were previously reported to be associated with inflammatory response. In this regard, Drackley [40] and Contreras and Sordillo [41] have proposed that inflammation is the missing link in the pathology of metabolic disorders in transition cows. Intense lipid metabolism, during the transition period, results in significant release of non-esterified fatty acids (NEFA) into the blood stream which can directly activate NF-ĸB signaling pathways [41, 42]. The NF-ĸB can induce the expression of several cytokines, chemokines and their receptors and subsequently enhance the cellular inflammatory response [42]. The metabolic effects of the acute systematic inflammation in transition cow results in mobilization of more adipose tissue, liver glycogen breakdown [43] and accumulation of the triglyceride in the liver [42]. All of these events are associated with ketosis and fatty liver in dairy cattle [44]. Taking all these results together, the authors of this study speculate that this region on BTA6 and the associated candidate genes might regulate pathways and events related to energy metabolism, inflammatory response function and cellular crosstalk in subclinical ketosis.
The GWAS results also showed three significant SNPs (BOVINEHD1000005959: rs110237995; BOVINEHD1000005943: rs110139536 and BOVINEHD1000005952: rs135236614, FDR < 5%) located on BTA10 at around 17 Mb (Additional file 1). Parker et al. (2018) reported a region on chromosome 10 associated with ketosis susceptibility; however, the position of this region was different from what was found in the current study. Three significant SNPs (FDR < 5%, Additional file 1) on chromosome 20 spanning around 55.3–57.4 Mb were also identified significant in this study. Two of these SNPs in this region (BOVINEHD2000015856: rs133951346; BOVINEHD2000015861: rs135815835) were located within the intronic region of gene F-box and leucine rich repeat protein 7 (FBXL7). The FBXL7 gene includes one of the four subunits of the E3 ubiquitin protein ligases, which play a role in phosphorylation-dependent ubiquitination of proteins (Gene ID: 23194). The ubiquitin system plays a pivotal role in the regulation of immune response and controls the basic aspect of the immune system, including lymphocyte development, differentiation and activation [45, 46]. The FBXL7 gene has also been reported to be located within a region that explains the highest genetic variance associated with clinical mastitis in first lactating US Holstein dairy cattle [47]. Therefore, this gene might be associated with inflammation factors that affect both clinical mastitis and clinical ketosis during first lactation in dairy cattle.
The IPA network enrichment analysis of the genes identified for SCK1 and their direct and indirect interactions with other molecules and genes are shown in Fig. 3. This network shows the association of most of the identified genes for SCK1 with two hub genes, IFNG and TNF. The IFNG gene encodes a soluble cytokine that is a member of type II interferon class and has important immune-regulatory functions in both innate and adaptive immune systems (Gene ID: 3458). In addition, cows with metritis or clinical endometritis show higher serum concentrations of leptin, adiponectin, IL-1β, IL-6and TNF-α [48]. In transition dairy cows, once mobilized NEFA reach the liver, the TNF-α decreases liver glucose production [43] and increase the triglyceride accumulation which can promote metabolic disorders [42]. Kasimanickam et al. [48] also reported that an increase in anti- and pro-inflammatory cytokines early in lactation is associated with uterine postpartum pro-inflammatory conditions, increase in adipose tissue metabolism and loss of body condition score; these last two factors are key biological features that affect ketosis and subclinical ketosis in dairy cattle [28, 29, 48].
Candidate SNP enrichment analysis for SCK1 has resulted in enrichment of seven significant GO terms (FDR ≤ 1%), introducing 71 SNPs as candidate SNPs (Additional file 2). From this significant GO terms, several terms including GO:0051180 (vitamin transport), GO:0051183 (vitamin transmembrane transport activity) and GO:0042359 (vitamin D metabolic process) were related to vitamin transport within and between cells and chemical reactions and pathways involving vitamin D (Additional file 2). Vitamin D has been reported to have regulatory effects on adipose tissue, lipid storage and calcium metabolism in humans and it has been shown that vitamin D receptor (VDR) is expressed in adipose tissue [49]. It has also been reported that plasma 25-hydroxy-vitamin D3(25OHD3) is inversely related to metabolic events and obesity complications, including hyperglycemia, hypertension, insulin resistance and dyslipidemia in humans [50].
Subclinical ketosis in second and later lactations (SCK2)
A total number of 369 SNPs (FDR < 5%) were identified significant for SCK2 in this study. These SNPs were mainly located on BTA6, 14 and 20 (Fig. 1b). The list of all significant SNPs and chromosomes are given in the Additional file 1.
The presence of these significant regions on chromosomes (6, 14 and 20) for SCK2 was reported in a previous short paper by Nayeri et al. [51] where the largest peak was located on BTA14 (Fig. 1b). In the following, the position of significant SNPs, their functional associations and other identified significant regions will be further discussed. One highly significant SNP on BTA14 (ARS-BFGL-NGS-4939: rs109421300, FDR < 1%) was shown to be an intronic SNP within DGAT1 gene (Additional file 1). Association of the DGAT1 gene with milk fat content [52, 53] and several production traits has been reported in several previous studies [53,54,55]. However, there were no previous indications of association of the identified SNP in this region with ketosis in previous studies. The enzyme produced by the DGAT1 gene (acyl-CoA: diacylglycerol acyltransferase) catalyzes the last step of triacyglyceride (TAG) synthesis in the liver and can significantly influence the energy metabolism in dairy cows [56]. It has been shown that excessive accumulation of TAG in liver and oxidative stress as a result of altered lipid metabolism early in lactation can increase the risk of metabolic disease including fatty liver and ketosis in dairy cattle [56]. Estimates of genetic correlations between 305-d milk yield and ketosis were found to be mostly unfavorable, with estimates ranging from 0.00 to 0.77 [57,58,59]. Therefore, one would expected QTL/genes with pleiotropic effects on both ketosis and milk yield would exist. This seems to be observed in this study, but the mechanism of the pleiotropic effects might involve intricate indirect pathways. These findings may also support the IPA enrichment analysis results, in which the DGAT1 enzyme was shown to be interacting indirectly with LEP and TNF genes (Fig. 4). Leptin is a hormone produced in the adipose tissue and its function is mainly to maintain glucose homeostasis and regulate appetite and energy metabolism in dairy cattle [60]. The TNF gene encodes a pro-inflammatory cytokine that is mainly secreted by macrophages; this cytokine, as explained previously, is involved in regulation of different biological processes including cell differentiation, proliferation, apoptosis and lipid metabolism and has been implicated in different autoimmune and metabolic-associated diseases including insulin resistance (Gene ID: 7124). Investigations on human and various animal models suggest that fatty acids can affect the host inflammatory responses in several ways [61]. One way is through the altered lipid metabolism, which often results in the increased levels of NEFA in the blood. Fatty acids are key resources of energy metabolism and are oxidized to produce Acyl-CoA and ATP. When there is a nutrient surplus, adipose tissue stores a large quantity of fatty acids in the form of TAGs [3]. This stored fatty acids can be then mobilized by lipolysis when there is an energy deficit, including early in lactation in dairy cattle [62, 63], which leads to increased NEFA in the blood for uptake by various tissues [64]. However, other tissues and cells may not be able to handle a sustained nutrient overload which might result in adverse effects [63]. Increased levels of circulating NEFAs are associated with increased systemic inflammatory conditions in humans [63]. Excessive fat mobilization due to a severe energy deficit impairs the cow’s immune function and fertility and leads to metabolic stress [15]. Therefore, altered lipid metabolism, increased concentrations of non-esterified fatty acids, excessive adipose stores and oxidative stress during the onset of lactation in dairy cattle, may negatively affect the inflammatory response of the individual to several pro-inflammatory diseases including metritis, mastitis and ketosis. This might explain the underlying biological pathways and interactions of the genes DGAT1, LEP and TNF and their associations with subclinical ketosis.
Interestingly, in this study the GWAS identified two highly significant SNPs (BovineHD4100010548: rs110651119; BovineHD1400000400: rs109221516, FDR < 1%) at around 25 Mb on BTA14 located in the gene lymphocyte antigen 6 family member H (LY6H), and two significant SNPs (BovineHD1400000443: rs133518905; BovineHD1400000446: rs136122630 at FDR < 1% and < 5%, respectively) at around 27 Mb located in the gene lymphocyte antigen 6 family member K (LY6K) (Table 2). The putative role of these two genes (LY6H and LY6K) in immune response has been reported in several previous investigations [65,66,67] and LY6K gene was reported to be a significant candidate gene for mastitis susceptibility in US and Chinese Holstein cattle [47, 68]. Therefore, these regions, underlying genes and biological pathway might be also associated with subclinical ketosis in later lactations.
Two other highly significant peaks for SCK2 were located on BTA6 (spanning around 78.7–98.6 Mb) and BTA20 (55–63 Mb) (Fig. 1b, Additional file 1). Three SNPs on BTA20 (at around 53 and 57 Mb) and a region on BTA6 (spanning around 88~93 Mb) were found to overlap between SCK1 and SCK2. One significant intronic SNP (BovineHD4100005290: rs110899052, FDR < 5%) on BTA6 at 87 Mb in this study was located in the gene casein alpha s1 (CSN1S1). Associations of two significant SNPs within the promotor region of the CSN1S1 gene with milk yield traits in German Angeln dairy cattle were reported in a quantitative trait loci (QTL) mapping study performed by Sanders et al. (2006) [69]. The presence of the significant genomic regions near the casein gene cluster (at 87 Mb) affecting milk production and milk component traits has been reported in several breeds including North American, Dutch, and US Holstein cattle for milk fat, milk protein yield and protein deviations [53, 70], in German Holstein [71] and Brazilian Holstein cattle [72] for milk yield and fat yield; however, this region was not reported to be associated with ketosis in previous studies. This region and its related gene might be associated with the high fat:protein ratio and decrease in milk protein percentage in early lactation [73].
Four SNPs in the current study (BovineHD1100004827: rs110571223; BovineHD1100004831: rs43672311) within gene BIRC6 and (BovineHD1100004868: rs110243275; BovineHD1100004913: rs42050566) within the gene TTC27 at 15~15.3 Mb were significant (FDR < 5%) (Additional file 1) on BTA11. This result is in agreement with the result of a GWAS study on ketosis susceptibility in Jersey dairy cows, in which significant regions were reported on BTA11 between 14.9 to 16.9 [29]. This region was reported to be associated with genes and pathways that regulate several functions, including metabolism, mastitis, steroid hormone metabolism and innate immune system receptors [74, 75]. Several SNPs on BTA25, spanning around 25.1–27.9 Mb for SCK2 (Additional file 1) were also identified. This result is supported by a gene-based mapping analysis of metabolic adaptation and metabolic disease in dairy cattle, in which two genes (ENSBTAG00000031551: PRSS53; ENSBTAG00000000781: HIP1) at around 27 Mb on BTA25 were shown to be associated with blood BHB concentrations and metabolic disorders [1]. Moreover, one highly significant SNP identified in the current study (BovineHD2500007432: rs42072596, FDR < 1%) on BTA25 was previously reported to be associated with carnitine and glycerophosphocholine in milk of Danish Holstein cattle [76]. This SNP is an intron variant located within the gene CLN3 which encodes a protein involved in lysosomal function (Gene ID: 1201). Mutations in this gene are associated with a neurodegenerative disease in humans which leads to reduced levels of carnitine in plasma [77]. Carnitine has been an important component of mitochondria involved in fatty acid β-oxidation and lipid metabolism [78]. Additionally, Buitenhuis et al. [31] reported a high correlation between milk carnitine and choline (0.86), and choline and BHB in milk (0.97). Moreover, Klein et al. (2011) showed that high values of glycerophosphocholine in milk throughout the lactation period are connected with a low ketosis incidence in dairy cattle [79]. Therefore, this significant SNP and its assigned gene (CLN3) might have an important biological function associated with subclinical ketosis and other correlated metabolic traits in dairy cattle. Another highly significant SNP (BovineHD2500007435: rs42071217, FDR < 1%) at around 26 Mb on BTA25 within the candidate gene APOBR was also identified in the current study, which was previously reported to carry a causative variant highly associated with milk levels of glycerophosphocholine in German Holstein dairy cattle [15]. Other putative functionally important candidate genes for SCK2 on chromosome 25 found in this study were IL4R (BovineHD2500007120: rs42062457), and PSPH (BovineHD2500007818: rs109700166) genes.
The significant peak found on BTA20 for SCK2 was located at 55.3~63.9 Mb. Several significant SNPs (FDR < 1%) on this region (32 SNPs) were located within or very close to the gene trio Rho guanine nucleotide exchange factor (TRIO) (Table 2). This gene encodes a large protein that functions as a GDP to GTP exchange factor (Gene ID: 7204) and was reported to be a significant candidate gene for maternal effect on weaning weight and milk yield traits in Blonde d’Aquitaine beef cattle [80]. It has also been shown that the TRIO gene controls leukocyte trans-endothelial migration during inflammatory conditions and other diseases, such as rheumatoid arthritis [81]. Thus, this significant region on chromosome 20 and its associated gene might be also related to the inflammatory response in transition dairy cow and the occurrence of metabolic disorders. Other positional candidate genes that were identified in this study on BTA20 (55–63 Mb) were ANKH, MYO10 and DNAH5. This region (at 55–63 Mb) on BTA20, was not reported to be associated with ketosis or subclinical ketosis in previous studies, therefore this region seems to be a novel region for subclinical ketosis.
Of the 369 SNPs that were used as candidate SNPs (FDR < 5%) in SNP2GO for enrichment analysis, a total of 114 SNPs were overrepresented in 91 enriched GO terms (Additional file 2). Some of these terms showed a clear association with SCK2 including GO:0030073 (insulin secretion); GO:0001678 (cellular glucose homeostasis) and GO:0046883 (regulation of hormone secretion). In order to have a better insight of the key biological pathways and their association with subclinical ketosis, GO terms were summarized and visualized using REVIGO web-server (Fig. 5). Blue and green bubbles show the GO terms with more significant P-values. The size of the bubbles indicates the frequency of the GO terms. One important GO term in the scatterplot is a term associated with regulation of Rho protein signal transduction; many researches over the last decade have uncovered the molecular links between the RhoGTPase and the NFKB pathways, and its association with inflammation and immune response [82].
Overlapping regions among SCK1 and SCK2
The only overlapping peaks between the SCK1 and SCK2 traits were in the regions identified on chromosome 6 at 88–93 Mb and chromosome 20 at 57 Mb. These regions affect milk BHB concentrations (subclinical ketosis) both in first and later lactations in cows. In dairy cattle milk production increases in later parities [83]. Production of more milk might be associated with increased milk fat content due to adipose mobilization and a higher fat:protein ratio [84].
Conclusions
The genome-wide association analysis on MIR predicted BHB milk in this study identified several significant regions associated with subclinical ketosis in first (SCK1) and later (SCK2) lactations in North American Holstein dairy cattle. The significant regions were mostly located on BTA6, 14 and 20. Regions on BTA6 and BTA20 were found to overlap between SCK1 and SCK2. Several of the identified regions were reported in previous investigations as associated with ketosis biomarkers, milk production, clinical mastitis and immunity in dairy cattle. However, a novel region on BTA20 at 55–63 Mb was also identified, which was not previously reported. Functional analysis identified pathways and biological terms and processes involved in lipid metabolism and immune function. The results of this study can be used for further genetic analysis to identify genes and causal variants that affect ketosis and other correlated metabolic diseases.
Methods
Animals and data
The Canadian Dairy Network (CDN, Guelph, Ontario, Canada) provided pedigree, genotypes and estimated breeding values (EBV) of cows and proven bulls for MIR predicted milk BHB in first lactation (SCK1) and later lactations (SCK2). Milk BHB measurements have been collected as a part of routine phenotyping via MIR spectroscopy by Canadian DHI since January 2013 using an existing FOSS calibration model and MilkoScan FT+ (FOSS Analytical A/S, Hillerød, Denmark) [85]. The correlation between chemical methods for measuring milk BHB and predictions using MIR spectroscopy has been shown to be approximately 0.80 [27].
Genotypes of 24,657 Canadian Holstein bulls (9,856) and cows (14,801) with the BovineSNP50K (50 K, 41,097 SNPs) BeadChip (Illumina, San Diego, CA) were provided by CDN. The SNPs in the 50 K panel passed standard quality control measures used by CDN. These genotypes were then imputed to the high density (HD, 311,725 SNPs, after editing 777 K panel for redundant SNPs [86]) genotypes (with a reference population of 2,507 animals) using FImpute V2.2 software [87]. Quality control was performed on the imputed HD genotyping data using the snp1101 software [88] and SNPs with low call rate < 0.9, low minor allele frequency (MAF) < 0.01 and excess of heterozygosity > 0.15 were excluded. After quality control, a total of 298,210 SNPs remained which were used for further genome-wide association analysis.
Estimation of breeding values for milk BHB
Estimated breeding values were calculated by CDN with a multiple-trait (9 traits) linear animal model [89] including metabolic disease traits, milk recording indicators, and body condition score. Milk BHB, or sub-clinical ketosis, for first and later (up to fifth) lactations is recorded at the first test-day from 5 to 45 days in milk (DIM), expressed as milk BHB in mmol/L and log transformed. Data for lactations greater than 2 are treated as repeated observations. The model equation for milk BHB at first lactation included the fixed effects of herd, year-season, DIM, age-season-parity, and random herd-year and animal additive genetic effects. For later lactations, a random permanent environment effect was added to account for repeated measures. Specific model details and assumptions can be found in Jamrozik et al. (2016).
Calculating de-regressed EBV
The EBV for MIR predicted milk BHB were de-regressed as explained in Garrick et al. [90] to be used in the genome-wide association study (GWAS). The Garrick et al. (2009) regression procedure adjusts for ancestral information and eliminates shrinkage contained in the EBV; therefore, the de-regressed EBV only contains their own and the descendant’s information on each animal [90].
Genome wide association study
The association analysis was performed using a single SNP regression mixed linear model implemented in the snp1101 software [91]. The mixed linear model used in this study was as below: Yi = μ + βgi + ai + ei.
where Yi is pseudo phenotype of the ith bull (bull’s de-regressed EBV); μ is the overall mean; β is the linear regression coefficient (allele substitution effect) of the SNP; gi is the SNP genotype of the ith bull (coded as 0 for BB, 1 for AB and 2 for AA SNP genotypes); ai is the random additive polygenic effect of the ith bull and ei is the random residual effect.
Assumptions of the model include a~N(0, Gσ a2 ) in which G is the genomic relationship matrix [91] and σ a2 is the polygenic additive genetic variance; e ~N(0, Rσ e2) in which σ e2 is the residual variance, where a and e are vectors of additive polygenic and residual effects, respectively. R is a diagonal matrix containing weights for the residual variance based on the reliabilities of the de-regressed bull EBV [91].
In order to account for multiple testing, genome-wise false discovery rate (FDR) of 5 and 1% were used to identify significant and highly significant associations, respectively. The inflation factor λ [91] and quantile-quantile (Q-Q) plots were also calculated to compare observed distributions of –log(P-value) to the expected distribution under the no association model for each trait.
Enrichment analysis
Significant SNPs at the level of 5% FDR were mapped to the corresponding genes in Ensemble database (Ensembl 90, Bos taurus UMD3.1, http://useast.ensembl.org/Bos_taurus/Info/Index) using the getBM() function in R-biomaRt package (https://www.bioconductor.org/) [92]. Genes within ±100 kb from the identified significant SNPs were used for further functional analysis.
Functional analysis was performed to identify the biological pathways and gene-networks associated with the list of positional candidate genes using the Ingenuity Pathway Analysis software (IPA; Ingenuity System Inc., USA). Additionally, a candidate SNP enrichment analysis was performed to identify genomic annotations and associated Gene Ontology terms (GO terms) for each trait using SNP2GO R package, as explained by Szkiba et al. (2014). For the GO term enrichment analysis, the Ensembl version 90 genomic annotation file (for Bos taurus UMD 3.1 assembly) in conjugation with Ensembl gene ID file (from Ensembl version 91 with gene ID and GO term accession) was used. The SNP2GO “extension” and the “runs” parameters were set to 50 nucleotides and 100,000, respectively and a false discovery rate (FDR) of 1% was used to correct for multiple testing [93]. The collection of enriched GO terms (FDR < 0.01) resulted from the SNP2GO analysis were then summarized and visualized using REVIGO web server (http://revigo.irb.hr) [94, 95]. This analysis assists the interpretation of the result by reducing the number of redundant enriched GO terms, using a simple clustering algorithm, and produces a scatterplot, relying on semantic similarities [95]. For this analysis, the GO terms enriched for both traits and the P-values from SNP2GO were uploaded to REVIGO. Settings used for REVIGO program were: database: Bos taurus, semantic similarity: 0.5 (small), semantic similarity measure: SimRel.
Abbreviations
- BHB:
-
β-hydroxybutyric acid
- BHBA:
-
β-hydroxybutyric acid
- BTA:
-
Bos taurus autosome
- CDN:
-
Canadian dairy network
- FDR:
-
false-discovery rate
- GO:
-
gene ontology
- GWAS:
-
genome-wide association study
- IPA:
-
ingenuity pathway analysis
- MIR:
-
Mid-infrared spectroscopy
- NEFA:
-
non-esterified fatty acid
- Q-Q plots:
-
Quantile-Quantile plots
- QTL:
-
quantitative trait loci
- SCK1:
-
subclinical ketosis in first lactation
- SCK2:
-
subclinical ketosis in second lactation and after
- SNP:
-
single nucleotide polymorphism
References
Ha N-T, Gross JJ, van Dorland A, Tetens J, Thaller G, Schlather M, et al. Gene-Based Mapping and Pathway Analysis of Metabolic Traits in Dairy Cows. PLoS One. 2015;10:e0122325. https://doi.org/10.1371/journal.pone.0122325.
Kessel S, Stroehl M, Meyer HHD, Hiss S, Sauerwein H, Schwarz FJ, et al. Individual variability in physiological adaptation to metabolic stress during early lactation in dairy cows kept under equal conditions. J Anim Sci. 2008;86:2903–12.
Nayeri S, Stothard P. Tissues, Metabolic Pathways and Genes of Key Importance in Lactating Dairy Cattle. Springer Sci Rev. 2016;4:49–77.
Le Blanc S. Monitoring metabolic health of dairy cattle in the transition period. J Reprod Dev. 2010;56 Suppl:S29–S35. https://doi.org/10.1262/jrd.1056S29.
Suthar VS, Canelas-Raposo J, Deniz A, Heuwieser W. Prevalence of subclinical ketosis and relationships with postpartum diseases in European dairy cows. J Dairy Sci. 2013;96:2925–38. https://doi.org/10.3168/jds.2012-6035.
Pryce JE, Parker Gaddis KL, Koeck A, Bastin C, Abdelsayed M, Gengler N, et al. Invited review: Opportunities for genetic improvement of metabolic diseases. J Dairy Sci. 2016;99:6855–73. https://doi.org/10.3168/jds.2016-10854.
Kadarmideen HN. Genomics to systems biology in animal and veterinary sciences: Progress, lessons and opportunities. Livest Sci. 2014;166:232–48. https://doi.org/10.1016/j.livsci.2014.04.028.
Koeck A, Jamrozik J, Kistemaker GJ, Schenkel FS, Kelton DF, Miglior F. Development of Genetic Evaluations for Metabolic Disease Traits for Canadian Dairy Cattle. 2015;:76–9.
Koeck A, Heringstad B, Egger-Danner C, Fuerst C, Fuerst-Waltl B. Comparison of different models for genetic analysis of clinical mastitis in Austrian Fleckvieh dual-purpose cows. J Dairy Sci. 2010;93:4351–8. https://doi.org/10.3168/jds.2009-2955.
Radostits O, Gay C, Hinchcliff K, Constable P. Veterinary Medicine: A Textbook of the Diseases of Cattle, Horses, Sheep, Pigs and Goats, 10th edition Radostits. 10th ed. Philadelphia, PA: Saunders Elsevier; 2010. https://doi.org/10.1111/j.1911-3846.1997.tb00533.x.
Biswal S, Nayak DC, Sardar KK. Prevalence of ketosis in dairy cows in milk shed areas of Odisha state ,. 2016;9:1242–7.
Koeck A, Jamrozik J, Schenkel FS, Moore RK, Lefebvre DM, Kelton DF, et al. Genetic analyses of milk B-hydroxybutryrate and its association with fat-to-protein ratio, body condition score, clinical ketosis, and displaced abomasum in early first lactation of Canadian Holsteins. J Dairy Sci. 2014;97:7286–92. https://doi.org/10.3168/jds.2014-8405.
Duffield TF, Lissemore KD, Mcbride BW, Leslie KE. Impact of hyperketonemia in early lactation dairy cows on health and production. J Dairy Sci. 2009;92:571–80. https://doi.org/10.3168/jds.2008-1507.
Oetzel GR. Understanding the Impact of Subclinical Ketosis. 2015;:15–26.
Tetens J, Heuer C, Heyer I, Klein MS, Gronwald W, Junge W, et al. Polymorphisms within the APOBR gene are highly associated with milk levels of prognostic ketosis biomarkers in dairy cows. 2015;:129–37.
Jorritsma R, Baldeé SJC, Schukken YH, Wensing TH, Wentink GH. Evaluation of a milk test for detection of subclinical ketosis. Vet Q. 1998;20:108–10.
McArt JAA, Nydam DV, Oetzel GR. Epidemiology of subclinical ketosis in early lactation dairy cattle. J Dairy Sci. [Internet]. Elsevier; 2012;95:5056–66. http://linkinghub.elsevier.com/retrieve/pii/S002203021200519X.
Raboisson D, Mounié M, Maigné E. Diseases, reproductive performance, and changes in milk production associated with subclinical ketosis in dairy cows: A meta-analysis and review. J Dairy Sci. [Internet]. Elsevier; 2014;97:7547–63. http://linkinghub.elsevier.com/retrieve/pii/S0022030214006912.
Fønss A, Munksgaard L. Automatic blood sampling in dairy cows. Comput Electron Agric. 2008;4:27–33.
van der Drift SGA, van Hulzen KJE, Teweldemedhn TG, Jorritsma R, Nielen M, Heuven HCM. Genetic and nongenetic variation in plasma and milk β-hydroxybutyrate and milk acetone concentrations of early-lactation dairy cows. J Dairy Sci. 2012;95:6781–7.
Denis-Robichaud J, Dubuc J, Lefebvre D, Des Côteaux L. Accuracy of milk ketone bodies from flow-injection analysis for the diagnosis of hyperketonemia in dairy cows. J Dairy Sci. [Internet]. Elsevier; 2014;97:3364–70. http://linkinghub.elsevier.com/retrieve/pii/S0022030214002100.
Belay TK, Dagnachew BS, Kowalski ZM, Ådnøy T. An attempt at predicting blood β-hydroxybutyrate from Fourier-transform mid-infrared spectra of milk using multivariate mixed models in Polish dairy cattle. J Dairy Sci. 2017;100:6312–26. https://doi.org/10.3168/jds.2016-12252.
De Marchi M, Toffanin V, Cassandro M, Penasa M. Invited review: Mid-infrared spectroscopy as phenotyping tool for milk traits1. J Dairy Sci. 2014;97:1171–86. https://doi.org/10.3168/jds.2013-6799.
Barbano DM, Lynch JM. Major Advances in Testing of Dairy Products : Milk Component and Dairy Product Attribute Testing. J Dairy Sci. 2006;89:1189–94.
Jamrozik J, Koeck A, Kistemaker GJ, Miglior F. Multiple-trait estimates of genetic parameters for metabolic disease traits, fertility disorders, and their predictors in Canadian Holsteins. J Dairy Sci. [Internet]. Elsevier; 2016;99:1990–8. http://linkinghub.elsevier.com/retrieve/pii/S0022030216000461.
Grelet C, Bastin C, Daviere J, Mohan M, Werner A, Reding R, et al. Development of Fourier transform mid-infrared calibrations to predict acetone, B-hydroxybutryrate, and citrate contents in bovine milk through a European dairy network. J Diary Sci. 2016;99:4816–25.
McCabe M, Waters S, Morris D, Kenny D, Lynn D, Creevey C. RNA-seq analysis of differential gene expression in liver from lactating dairy cows divergent in negative energy balance. BMC Genomics. [Internet]. 2012;13:193. Available from: http://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-13-193.
Hillreiner M, Flinspach C, Pfaffl MW, Kliem H. Effect of the Ketone Body Beta- Hydroxybutyrate on the Innate Defense Capability of Primary Bovine Mammary Epithelial Cells. Plus One. [Internet]. 2016;11:1–18. Available from: https://doi.org/10.1371/journal.pone.0157774.
Parker Gaddis KL, Megonigal JH Jr, Clay JS, Wolfe CW. Genome-wide association study for ketosis in US Jerseys using producer-recorded data. J Dairy Sci. 2018;101:413–24.
Chaudhari A, Håversen L, Mobini R, Andersson L, Ståhlman M, Lu E, et al. Biochimica et Biophysica Acta ARAP2 promotes GLUT1-mediated basal glucose uptake through regulation of sphingolipid metabolism. Biochim Biophys Acta. Elsevier B.V.; 2016;1861:1643–51.
Buitenhuis AJ, Sundekilde UK, Poulsen NA, Bertram HC, Larsen LB, Sørensen P. Estimation of genetic parameters and detection of quantitative trait loci for metabolites in Danish Holstein milk. J Dairy Sci. [Internet]. Elsevier; 2013;96:3285–95. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0022030213001859.
Mömke S, Scholz H, Doll K, Rehage J, Distl O. Mapping quantitative trait loci for left-sided displacement of the abomasum in German Holstein dairy cows. J Dairy Sci. 2008;91:4383–92. https://doi.org/10.3168/jds.2008-1260.
Wathes DC, Cheng Z, Chowdhury W, Fenwick MA, Fitzpatrick R, Morris DG, et al. Negative energy balance alters global gene expression and immune responses in the uterus of postpartum dairy cows. 2009;:1–13.
Esposito G, Irons PC, Webb EC, Chapwanya A. Interactions between negative energy balance , metabolic diseases , uterine health and immune response in transition dairy cows. Anim Reprod Sci. 2014;144:60–71. https://doi.org/10.1016/j.anireprosci.2013.11.007.
Naeem A, Drackley JK, Stamey J, Loor JJ. Role of metabolic and cellular proliferation genes in ruminal development in response to enhanced plane of nutrition in neonatal Holstein calves 1. J Dairy Sci. 2012;95:1807–20. https://doi.org/10.3168/jds.2011-4709.
Olsen HG, Knutsen TM, Sabat AML, Grove H, Nome T, Svendsen M, et al. Fine mapping of a QTL on bovine chromosome 6 using imputed full sequence data suggests a key role for the group - specific component (GC) gene in clinical mastitis and milk production. Genet Sel Evol. 2016;48:1–16.
Nelson CD, Reinhardt TA, Thacker TC, Beitz DC, Lippolis JD. Modulation of the bovine innate immune response by production of 1α,25-dihydroxyvitamin D3 in bovine monocytes. J Dairy Sci. [Internet]. Elsevier; 2010;93:1041–9. http://linkinghub.elsevier.com/retrieve/pii/S0022030210000718.
Nelson C, Lippolis J. Activation of Vitamin D3 in Bovine Mastitis Caused by Streptococcus uberis. 2009;655:1–3.
Sorge US, Molitor T, Linn J, Gallaher D, Wells SW. Cow-level association between serum 25-hydroxyvitamin D concentration and Mycobacterium avium subspecies paratuberculosis antibody seropositivity: A pilot study. J Dairy Sci. [Internet]. Elsevier; 2013;96:1030–7. Available from: http://linkinghub.elsevier.com/retrieve/pii/S002203021200923X.
Drackley JK. Biology of dairy cows during the transition period: the final frontier? J Dairy Sci. [Internet]. Elsevier; 1999;82:2259–73. https://doi.org/10.3168/jds.S0022-0302(99)75474-3.
Contreras GA, Sordillo LM. Comparative Immunology, Microbiology and Infectious Diseases Lipid mobilization and inflammatory responses during the transition period of dairy cows. Comparative Immunol Microbiol Infect Dis. [Internet]. Elsevier Ltd; 2011;34:281–9. https://doi.org/10.1016/j.cimid.2011.01.004.
Garcia-Ruiz I, Rodríguez-Juan C, Díaz-Sanjuan T, Hoyo P, Colina F, Munoz-Yague T, et al. Uric Acid and Anti-TNF Antibody Improve Mitochondrial Dysfunction in ob/ob Mice. Hepatology. 2006;44:581–91.
Kettelhut IC, Fierst W, Goldberg AL. The toxic effects of tumor necrosis factor in vivo and their prevention by cyclooxygenase inhibitors. Proc Natl Acad Sci USA. 1987;84 June:4273–4277.
Ingvartsen KL. Feeding- and management-related diseases in the transition cow Physiological adaptations around calving and strategies to reduce feeding-related diseases
 . Anim Feed Sci Technol. 2006;126:175–213.
. Anim Feed Sci Technol. 2006;126:175–213.Liu Y, Lear T, Zhao Y, Zhao J, Zou C, Chen BB, et al. F-box protein Fbxl18 mediates polyubiquitylation and proteasomal degradation of the pro-apoptotic SCF subunit Fbxl7. Cell Death Dis. 2015;6:e1630–9. Available from: https://doi.org/10.1038/cddis.2014.585.
Park Y, Jin H Seung, Aki D, Lee J, Liu YC. The ubiquitin system in immune regulation. 1st edition. Elsevier Inc.; 2014. https://doi.org/10.1016/B978-0-12-800147-9.00002-9.
Tiezzi F, Parker-gaddis KL, Cole JB, Clay JS. A Genome-Wide Association Study for Clinical Mastitis in First Parity US Holstein Cows Using Single-Step Approach and Genomic Matrix Re-Weighting Procedure. 2015;1–15. Available from: https://doi.org/10.1371/journal.pone.0114919.
Kasimanickam RK, Kasimanickam VR, Olsen JR, Jeffress EJ, Moore DA, et al. Associations among serum pro- and anti-inflammatory cytokines, metabolic mediators, body condition, and uterine disease in postpartum dairy cows. Reprod Biol Endocrinol. 2013;11:103. https://doi.org/10.1186/1477-7827-11-103.
Wamberg L, Kampmann U, Stødkilde-Jørgensen H, Rejnmark L, Pedersen SB, Richelsen B. Effects of vitamin D supplementation on body fat accumulation, inflammation, and metabolic risk factors in obese adults with low vitamin D levels - Results from a randomized trial. Eur J Intern Med. [Internet]. Elsevier B.V.; 2013;24:644–9. https://doi.org/10.1016/j.ejim.2013.03.005.
Forman JP, Curhan GC, Taylor EN. Plasma 25-hydroxyvitamin D levels and risk of incident hypertension among young women. Hypertension. 2008;52:828–32.
Nayeri S, Kroezen V, Sargolzaei M, Fleming A, Schenkel FS, Baes C, et al. Genome-wide Association Analysis for β-hydroxybutyrate Concentration in Milk Using Mid-Infrared Spectroscopy in North American Holstein Cattle. In: Proceedings of the World Congress on Genetics Applied to Livestock Production, 11.634. 2018. p. 1–5.
Grisart B, Coppieters W, Farnir F, Karim L, Ford C. Positional candidate cloning of a QTL in dairy cattle: identi cation of a missense mutation in the bovine DGAT gene with major effect on milk yield and composition. Genome Res. 2002;12:222–31.
Nayeri S, Sargolzaei M, Abo-Ismail MK, May N, Miller SP, Schenkel F, et al. Genome-wide association for milk production and female fertility traits in Canadian dairy Holstein cattle. BMC Genet. 2016;17:75. https://doi.org/10.1186/s12863-016-0386-1.
Meredith BK, Kearney FJ, Finlay EK, Bradley DG, Fahey AG, Berry DP, et al. Genome-wide associations for milk production and somatic cell score in Holstein-Friesian cattle in Ireland. BMC Genet. [Internet]. BioMed Central Ltd; 2012;13:21. [cited 24 Nov 2014]; Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3361482&tool=pmcentrez&rendertype=abstract.
Maxa J, Neuditschko M, Russ I, Förster M, Medugorac I. Genome-wide association mapping of milk production traits in Braunvieh cattle. J Dairy Sci. [Internet]. Elsevier; 2012;95:5357–64. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22916942.
Lešková L, Bauer M, Chrenek P, Lacková Z, Soročinová J, Petrovič V, et al. Detection of DGAT1 gene polymorphism and its effect on selected biochemical indicators in dairy cows after calving Loor et al. (2006) examined temporal gene expression in the liver of Holstein cows , and identified 85 genes with expression patterns tha. Acta Vet Brno. 2013;265–9.
Mantysarri E, Grohn Y. Clinical Ketosis: Phenotypic and Genetic Correlations Between Occurrences and with Milk Yield. J Dairy Sci. 1991;74:3985–93.
Simianer H, Solbu H, Schaeffer LR. Estimated Genetic Correlations Between Disease and Yield Traits in Dairy Cattle h r e f r o along w i t. J Dairy Sci. 1991;74:4358–65.
Uribe HA, Kennedy B, Martin S, Kelton D. Genetic Parameters for Common Health Disorders of Holstein Cows. J Dairy Sci. 1995;78:421–30.
Priyadarshini L, Yadav AK, Singh HS, Mishra A, Jain AK, Ahirwar MK. Role of leptin in physiology of animal reproduction-A review. Agric Rev. 2015;36:235–40.
Sordillo LM, Contreras GA, Aitken SL. Metabolic factors affecting the inflammatory response of periparturient dairy cows y. Anim Heal Res Rev. 2009;10:53–63.
Bauman DE, Griinari JM. Regulation and nutritional manipulation of milk fat: Low-fat milk syndrome. Livest Prod Sci. 2001;70:15–29.
Wood LG, Scott HA, Garg ML, Gibson PG. Progress in Lipid Research Innate immune mechanisms linking non-esterified fatty acids and respiratory disease. Prog Lipid Res. [Internet]. Elsevier Ltd; 2009;48:27–43. Available from: https://doi.org/10.1016/j.plipres.2008.10.001.
Roche JR, Friggens NC, Kay JK, Fisher MW, Stafford KJ, Berry DP. Invited review: Body condition score and its association with dairy cow productivity, health, and welfare. J Dairy Sci. [Internet]. Elsevier; 2009;92:5769–801. https://doi.org/10.3168/jds.2009-2431.
Paleari L, Catassi A, Ciarlo M, Cavalieri Z, Bruzzo C. Role of α 7-nicotinic acetylcholine receptor in human non-small cell lung cancer proliferation. Cell Prolif. 2008;936–59.
Zdanowski R, Krzyżowska M, Ujazdowska D, Lewicka A, Lewicki S. Role of α 7 nicotinic receptor in the immune system and intracellular signaling pathways. Cent Eur J Immun. 2015;40:373–9.
Mallya M, Campbell RD, Aguado B. Transcriptional Analysis of a Novel Cluster of LY-6 Family Members in the Human and Mouse Major Histocompatibility Complex: Five Genes with Many Splice Forms. Academic Press; Genomics. 2002;80:113–23. Available from: https://www.sciencedirect.com/science/article/pii/S0888754302967947?via%3Dihub. [cited 2 Jan 2018];
Wang X, Ma P, Liu J, Zhang Q, Zhang Y, Ding X, et al. Genome-wide association study in Chinese Holstein cows reveal two candidate genes for somatic cell score as an indicator for mastitis susceptibility. BMC Genet. [Internet]. 2015;16:111. Available from: http://www.biomedcentral.com/1471-2156/16/111.
Sanders K, Bennewitz J, Reinsch N, Thaller G, Prinzenberg E, Ku C, et al. Characterization of the DGAT1 Mutations and the CSN1S1 Promoter in the German Angeln Dairy Cattle Population. J Dairy Sci. 2006;89:3164–74.
Bovenhuis H, Van Arendonk JA, Korver S. Associations between milk protein polymorphisms and milk production traits. J Dairy Sci. 1992;75:2549–59.
Freyer G, Sørensen P, Kühn C, Weikard R, Hoeschele I. Search for pleiotropic QTL on chromosome BTA6 affecting yield traits of milk production. J Dairy Sci. 2003;86:999–1008.
Silva AA, Azevedo ALS, Verneque RS, Gasparini K, Peixoto MGCD, da Silva MVGB, et al. Quantitative trait loci affecting milk production traits on bovine chromosome 6 in zebuine Gyr breed. J Dairy Sci. [Internet]. Elsevier; 2011;94:971–80. Available from: https://doi.org/10.3168/jds.2009-2970.
Tetens J, Seidenspinner T, Buttchereit N, Thaller G. Whole-genome association study for energy balance and fat/protein ratio in German Holstein bull dams. Anim Genet. 2013;44:1–8.
Fabregat A, Sidiropoulos K, Garapati P, Gillespie M, Hausmann K, Haw R, et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2016;44:D481–7.
Guo H, Callaway JB, JPY T. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–87.
Buitenhuis B, Poulsen NA, Gebreyesus G, Larsen LB. Estimation of genetic parameters and detection of chromosomal regions affecting the major milk proteins and their post translational modifications in Danish Holstein and Danish Jersey cattle. BMC Genet. [Internet]. 2016;1–12. Available from: https://doi.org/10.1186/s12863-016-0421-2.
Katz ML. Decreased plasma carnitine and trimethyl-L-lysine levels associated with lysosomal accumulation of a trimethyl-L-lysine containing protein in Batten disease. Biochim Biophys Acta - Mol Basis Dis. 1996;1317:192–8.
McGarry JD, Brown NF. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur J Biochem. 1997;244:1–14.
Klein MS, Buttchereit N, Miemczyk SP, Immervoll AK, Louis C, Wiedemann S, et al. NMR metabolomic analysis of dairy cows reveals milk glycerophosphocholine to phosphocholine ratio as prognostic biomarker for risk of ketosis. J Proteome Res. 2012;11:1373–81.
Michenet A, Barbat M, Saintilan R, Venot E, Phocas F. Detection of quantitative trait loci for maternal traits using high-density genotypes of Blonde d’Aquitaine beef cattle. BMC Genet. 2016;17:1–13. https://doi.org/10.1186/s12863-016-0397-y.
van Rijssel J, Kroon J, Hoogenboezem M, van Alphen FPJ, de Jong RJ, Kostadinova E, et al. The Rho-guanine nucleotide exchange factor Trio controls leukocyte transendothelial migration by promoting docking structure formation. Mol Biol Cell. 2012;23:2831–44. https://doi.org/10.1091/mbc.E11-11-0907.
Tong L, Tergaonkar V. Rho protein GTPases and their interactions with NFκB: crossroads of inflammation and matrix biology. Biosci Rep. 2014;34:283–95. Available from: http://bioscirep.org/cgi/doi/10.1042/BSR20140021.
Guler O, Yanar M, Aydin R, Bayram B, Dogru U, Kopuzlu S. Genetic and Environmental Parameters of Milkability Traits in Holstein Friesian Cows. J Anim Vet Adv. 2009;8:143–7.
Buttchereit N, Stamer E, Junge W, Thaller G. Evaluation of five lactation curve models fitted for fat : protein ratio of milk and daily energy balance. J Dairy Sci. [Internet]. Elsevier; 2010;93:1702–12. https://doi.org/10.3168/jds.2009-2198.
Bonfatti V, Fleming A, Koeck A, Miglior F. Standardization of milk infrared spectra for the retroactive application of calibration models. American Dairy Science Association; J Dairy Sci. [Internet]. 2017;100:2032–41. http://linkinghub.elsevier.com/retrieve/pii/S0022030217300255.
Van Raden PM, Null DJ, Sargolzaei M, Wiggans GR, Tooker ME, Cole JB, et al. Genomic imputation and evaluation using high-density Holstein genotypes. J Dairy Sci. [Internet]. Elsevier; 2013;96:668–78. Available from: http://www.journalofdairyscience.org/article/S0022-0302(12)00757-6/abstract%5Cn, http://www.ncbi.nlm.nih.gov/pubmed/23063157. [cited 25 Oct 2014].
Sargolzaei M, Chesnais JP, Schenkel FS. A new approach for efficient genotype imputation using information from relatives. BMC Genomics. 2014;15:478. https://doi.org/10.1186/1471-2164-15-478.
Sargolzaei M. SNP1101 User’s Guide. Version 1.0. 2014;0.
Jamrozik J, Kistemaker GJ, Van Doormaal BJ, Fleming A, Koeck A, Miglior F. Genetic Evaluation for Resistance to Metabolic Diseases in Canadian Dairy Breeds. Interbull Bull. 2016;9–16.
Garrick DJ, Taylor JF, Fernando RL. Deregressing estimated breeding values and weighting information for genomic regression analyses. Genet Sel Evol. 2009;1:41:55.
Van Raden PM. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91:4414–23. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18946147. [cited 2014 Jul 12].
Durinck S, Spellman PT, Birney E, Huber W. Mapping identfiers for integration of genomic datasets with the R/Bioconductor package. biomaRt. 2009;4:1184–91.
Szkiba D, Kapun M, von Haeseler A, Gallach M. SNP2GO: functional analysis of genome-wide association studies. Genetics. [Internet]. 2014;197:285–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24561481. [cited 11 Dec 2014].
Schlicker A, Domingues FS, Rahnenführer J, Lengauer T. BMC Bioinformatics. 2006;16:1–16.
Supek F, Bosnjak M, Smuc T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. Plus One. 2011;6:1–9.
 . Anim Feed Sci Technol. 2006;126:175–213.
. Anim Feed Sci Technol. 2006;126:175–213.Acknowledgements
All dairy producers that participated in this project are gratefully acknowledged. We gratefully acknowledge Ian Rumbles (DRPC, Raleigh, NC) and the CanWest DHI team (Guelph, ON), Dr. Daniel Lefebvre and all the team members of Valecta (Ste-Anne-de-Bellevue,QC) for organizing the selection of herds and sample collecting for this project. We warmly acknowledge all the funding agencies.
Availability of the data and materials
Relevant information supporting the results not already presented in the article is given in additional files. The estimated breeding values and pedigree information are publicly available at CDN website (www.cdn.ca). The bulls’ genotypes cannot be made publicly available, because they are property of the bulls’ owners.
Funding
This study was partly funded by the DairyGen council of Canadian Dairy Network (Guelph, Ontario, Canada) and the Natural Sciences and Engineering Research Council of Canada (Ottawa, Ontario, Canada). This project is also partly supported by Agriculture and Agri-Food Canada and by additional contributions from Dairy Farmers of Canada, the Canadian Dairy Network and the Canadian Dairy Commission under the Agri-Science Clusters Initiative. This study received support from the Efficient Dairy Genome Project, funded by Genome Canada (Ottawa, Canada), Genome Alberta (Calgary, Canada), Ontario Genomics (Toronto, Canada), Alberta Ministry of Agriculture (Edmonton, Canada), Ontario Ministry of Research and Innovation (Toronto, Canada). Additional support was provided by the Ontario Ministry of Agriculture, Food and Rural Affairs (Guelph, Canada), GrowSafe Systems (Airdrie, Canada), Alberta Milk (Edmonton, Canada), Victoria Agriculture (Australia), Scotland’s Rural College (Edinburgh, UK), USDA Agricultural Research Service (United States), Qualitas AG (Switzerland), Aarhus University (Denmark).
Author information
Authors and Affiliations
Contributions
FM, FS, AF, CB, JS, AC and SN developed the project, assisted with data analysis and edited the manuscript. SN performed data analysis and drafted the manuscript. VK helped to interpret the result and AF help to editing of the manuscript. MS assisted with data analysis and edited the manuscript. All authors contributed to the manuscript and approved the final version of the manuscript.
Author’s information
Not Applicable
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Animal Care and Use Committee approval was not obtained for this study because analyses were performed on existing data obtained under standard farm management from commercial dairy farmers and breeders. Data used in this research was provided by CDN, which is the organization that runs the national dairy cattle genetic evaluations in Canada. All dairy farmers in Canada must follow “The Code of Practice for the Care and Handling of Dairy Cattle” developed by the National Farm Animal Care Council of Canada (http://www.nfacc.ca/).
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interest. The funders had no role in data analysis, decision to publish or preparation of the manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Excel file describing highly significant SNPs (genome-wise FDR < 5%) resulting from GWAS analysis using single SNP regression mixed linear model for subclinical ketosis in first lactation (SCK1) and subclinical ketosis in later lactations (SCK2) (XLS 91 kb)
Additional file 2:
Excel file describing gene ontology biological process terms (GO terms) enriched among GWAS results using SNP2GO enrichment analysis for subclinical ketosis in first lactation (SCK1) and subclinical ketosis in later lactations (SCK2) (XLSX 24 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Nayeri, S., Schenkel, F., Fleming, A. et al. Genome-wide association analysis for β-hydroxybutyrate concentration in Milk in Holstein dairy cattle. BMC Genet 20, 58 (2019). https://doi.org/10.1186/s12863-019-0761-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-019-0761-9