
Abstract
Background
One of the most commonly used quality measurements of pork is pH measured 24 h after slaughter. The most probable mode of inheritance for this trait is oligogenic with several known major genes, such as PRKAG3. In this study, we used whole-genome SNP genotypes of over 700 AI boars; after a quality check, 42,385 SNPs remained for association analysis. All the boars were purebred Finnish Yorkshire. To account for relatedness of the animals, a pedigree-based relationship matrix was used in a mixed linear model to test the effect of SNPs on pH measured from loin. A bioinformatics analysis was performed to identify the most promising genes in the significant regions related to meat quality.
Results
Genome-wide association study (GWAS) revealed three significant chromosomal regions: one on chromosome 3 (39.9 Mb–40.1 Mb) and two on chromosome 15 (58.5 Mb–60.5 Mb and 132 Mb–135 Mb including PRKAG3). A conditional analysis with a significant SNP in the PRKAG3 region, MARC0083357, as a covariate in the model retained the significant SNPs on chromosome 3. Even though linkage disequilibrium was relatively high over a long distance between MARC0083357 and other significant SNPs on chromosome 15, some SNPs retained their significance in the conditional analysis, even in the vicinity of PRKAG3. The significant regions harbored several genes, including two genes involved in cyclic AMP (cAMP) signaling: ADCY9 and CREBBP. Based on functional and transcription factor-gene networks, the most promising candidate genes for meat pH are ADCY9, CREBBP, TRAP1, NRG1, PRKAG3, VIL1, TNS1, and IGFBP5, and the key transcription factors related to these genes are HNF4A, PPARG, and Nkx2-5.
Conclusions
Based on SNP association, pathway, and transcription factor analysis, we were able to identify several genes with potential to control muscle cell homeostasis and meat quality. The associated SNPs can be used in selection for better pork. We also showed that post-GWAS analysis reveals important information about the genes’ potential role on meat quality. The gained information can be used in later functional studies.
Similar content being viewed by others

Background
The most common quality measurements of pork are pH and color. Both depend on post-mortem biochemical processes where lactate is produced from glycogen through glycogenolysis and anaerobic glycolysis, consequently lowering the pH and affecting the color of meat. The degree of glycolysis depends on the glycolytic potential, i.e. the amount of lactate that can be produced from glycogen in muscle at the time of slaughter [1]. Differences in glycolytic potential are known to be partly genetic. A well-known gene affecting meat quality is PRKAG3 (protein kinase, AMP-activated, gamma 3 non-catalytic subunit) [2], formerly known as the RN gene [3, 4]. An R200Q substitution in this gene is related to the upregulation of certain key enzymes (e.g. UDP-glucose pyrophosphorylase) that increase glycogen production in the muscle cells of 200Q animals [5]. Additionally, several other genes are associated with post-mortem pH and color of pork loin and ham, including RYR1 [6], CAST [7], and PHKG1 [8] among others.
Genome-wide association study (GWAS), based on abundant SNP markers, is an effective tool to find chromosomal regions that explain at least a moderate proportion of the genetic variance of the studied trait in a certain population. Since the launch of the commercial SNP chip for pigs (Illumina PorcineSNP60 Genotyping Beadchip), several GWAS for pork meat quality traits have been published [9–15]. These studies have detected significant associations between SNPs and pH on the following chromosomes: chromosome 14 [9]; 2, 3, 4, 13, and X [10]; 1, 2, 3, 7, 9, and 13 [11]; 2 and 15 [12]; 1 and 8 [13]; and 15 [14, 15].
The effective population size (Ne) of commercial pork breeds is relatively small; e.g. in Finnish Yorkshire, the pedigree- and marker-based estimate of Ne is only 60 animals [16]. This leads to strong linkage disequilibrium (LD) over long distances, and ultimately to a large number of genes surrounding the most significant SNP depending, of course, on how rich in genes the region is. Post-GWAS analysis, such as pathway and gene-transcription factor (TF) network analysis facilitate the identification and in silico validation of the most probable group of candidate genes in these regions and increases our understanding of the molecular mechanisms of the studied trait [17, 18].
In this article, we present the results from GWAS of loin pH measured 24 h post mortem using the estimated breeding values of Finnish Yorkshire boars. We also conducted a post-GWAS bioinformatics analysis of the significant genomic regions and genes.
Methods
Animals and phenotypes
The animals used in this study were AI boars of Finnish Yorkshire origin. The studied trait, pH of loin, was measured at the slaughterhouse 24 h post mortem using a Knick 752 pH meter and an Ingold 460 electrode (see Sevón-Aimonen et al. [19] for more information). The slaughtered pigs were grown in a test station. The animals arrived at the test station at 30 kg live weight and were sent for slaughter after 13 weeks of testing, at approximately 100 kg live weight. Prior to 2006, all pigs had restricted feeding based on their body weight; after 2006, feeding was close to ad libitum. The average pH for Finnish Yorkshire was 5.53 (SD = 0.15), based on 45,639 loin pH observations.
Estimated breeding values (EBVs) for the AI boars were calculated by Figen Oy (Seinäjoki, Finland) using a single-trait animal model and the Mix99 program package (MiX99, http://www.luke.fi/mix99. EBVs were mainly based on phenotypic measurements of half-sibs, full-sibs, and progeny. No phenotypic measurements of loin pH of the AI boars themselves were available. The statistical model included gender, slaughter batch, and time from slaughter to pH measurement as fixed effects, and litter and animal as random effects. Approximate reliabilities were calculated using ApaX, which is part of the MiX99 program package. All animals were related including, e.g., sire-son and half-sib pairs. Prior to association analysis, raw EBVs were deregressed and their weights calculated based on the method presented by Garrick et al. [20]. Deregressed EBVs (dEBVs) with weight less than 1.0 were removed from the data. The average reliability of the original EBVs was 0.56. The average dEBV was 0.09 and the SD 0.07, and the mean weight of the dEBVs was 3.6.
Genotypes
DNA was extracted from the boars either from hair follicles (boars born before 2008) or semen (boars born after 2008). Whole-genome SNP genotyping was done at FIMM (Institute for Molecular Medicine Finland, Helsinki) or at GeneSeek (Lincoln, NE, USA) using the Illumina PorcineSNP60 BeadChip (Illumina Inc., San Diego, CA, USA). Only animals with call rate (CR) >0.9 were included into the statistical analysis, 703 animals in total. Additionally, SNPs with CR <0.9, minor allele frequency (MAF) <0.1, and P-value of Hardy-Weinberg test statistics <0.00001 were removed from the data prior to the statistical analysis. The final data included 42,385 SNPs. The map positions of the SNPs were based on the genome build Sscrofa10.2.
Association analysis
Associations between loin pH and SNPs were tested for each SNP separately. Because the animals were related, the following linear model was applied to the data:
where y i is the deregressed EBV of an AI-boar i; μ is an intercept; b is the regression coefficient; and x i is the corresponding allele dose of the tested SNP with values of 0 (homozygous for the major allele), 1 (heterozygote), and 2 (homozygous for the minor allele); a i is the polygenic effect with a i ~ N(0, Aσ2 a); and e i is the residual effect with e i ~ N(0, Iσ2 e/w i ). Matrix A is the pedigree-based additive relationship matrix and I is the identity matrix with diagonal elements of σ2 e/w i , where weights (w i ) were calculated based on the reliabilities of the EBVs of the animal i itself and its parents. The SNP effect (b) and additive genetic (σ2 a) and residual (σ2 e) variances were estimated for each SNP separately using the Restricted Maximum Likelihood (REML) method of the DMU program package [21].
The statistical significance of the SNPs was based on a two-sided t-test with H0: b = 0, HA: b ≠ 0, and Bonferroni-corrected P-values. Because of the strong LD between the SNPs in Finnish Yorkshire [16], the number of independent tests is less than the number of SNPs in the data. Thus, in this research we used the previously defined P-value of 2.0E-06 based on the assumption of 25,000 independent tests, as a limit value for statistically significant association [22]. Haplotypes of the animals were estimated with FastPHASE [23] using default parameters, and Manhattan plots were produced with the Haploview program [24].
Post-GWAS analysis
For post-GWAS analysis, three lists of genes were formed based on genes within the significant regions: on chromosome 3 (39.4 Mb–40.1 Mb) and on chromosome 15 (58.5 Mb–60.5 Mb and 133.3 Mb–134.2 Mb). Additionally, we formed a separate list of genes relating to the significant SNPs on chromosome 15 outside the above regions, using 22.2 kb of the 5′ and 3′ flanking sequences (i.e. half the average distance between two SNPs on the chip). The annotations were based on the genome build Sscrofa10.2 at the NCBI website (https://www.ncbi.nlm.nih.gov/gene/). The two-sided hypergeometric test of the ClueGO plug-in of Cytoscape [25] was used to construct a gene network highlighting the pathways and relations across the four sets of genes.
The TFM-Explorer program (http://bioinfo.lifl.fr/TFM/TFME/) was used to identify the TF related to significant SNPs and regions. The TFM-Explorer takes a set of gene sequences and searches for locally overrepresented TF-binding sites (TFBS). The search protocol of the TFM-Explorer utilizes weight matrices from the JASPAR vertebrate database [26]. Our input into the program included 3,000 bp upstream and 300 bp downstream sequences of the gene transcription start sites in the FASTA format. Non-coding RNA genes were excluded from the analysis. The significance (P-value ≤ 0.01) of the clusters (regions of the input sequences associated with a factor) was based on a score function, as described by Touzet and Varré [27] and Defrance and Touzet [28].
The obtained list of TFs was analyzed for overrepresented gene ontology (GO) terms using the BiNGO (Biological Networks Gene Ontology tool, [29] plug-in of Cytoscape [30]). In the analysis, we applied the default statistical tests and corrections for multiple testing to retain an overall P-value of 0.05. A list of the most promising key TFs related to meat pH was formed based on the biological processes involved (e.g. muscle cell homeostasis) and a literature review. The most likely candidate genes were then identified using the NetworkAnalyzer tool within Cytoscape. Based on TFBS (and consequently, the number of connections in each gene and TF), the most connected genes were determined within each list of genes to form a gene-TF network.
Results
Significant SNPs
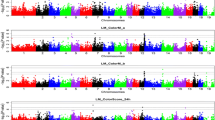
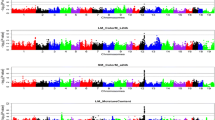
The Manhattan plots of GWAS are presented in Fig. 1. Three regions were statistically significant: one on chromosome 3 and two on chromosome 15. The allele effects with the standard errors and P-values of the significant SNPs are shown in Table 1 and the P-values (-log10) against the chromosomal positions with the validated genes from the Reference Sequence database (RefSeq, http://www.ncbi.nlm.nih.gov/refseq/) in Fig. 2.

Manhattan plots of P-values (-log10) from association analysis (a) and from conditional association analysis (b) of pH measured from loin

P-values (-log10-based) of the significant regions. a: chromosome 3, b and c: chromosome 15 against the positions of the validated genes
The statistically significant region on chromosome 3 was located at around 39.9 Mb–40.1 Mb and had four significant SNPs with an average allele effect of 0.018 pH units (SE = 0.004), corresponding to 0.4 SD of the total additive genetic effect and approximately 8% of the additive genetic variance (σa = 0.045 used in national breeding value evaluation for loin pH). In this region, the frequencies of unfavorable and favorable alleles were equal (MAF = 0.5). This region harbors three validated genes: TNF receptor-associated protein 1 (TRAP1), deoxyribonuclease I (DNASE1), and clusterin-associated protein 1 (CLUAP1).
The first region on chromosome 15 (58.5 Mb–60.5 Mb) had six statistically significant SNPs. The allele frequency of the minor allele of the best SNP (ASGA0069650) was 0.11 and the SNP effect was -0.031 pH units (SE = 0.006), corresponding to 0.7 SD of the total additive genetic effect and approximately 9% of the additive genetic variance. Only one gene, MAK16 homolog (MAK16), has been validated in this region (Fig. 2).
The second region on chromosome 15 (approximately 130 Mb–134 Mb) included PRKAG3, the gene reported earlier using partly the same data as here [12, 31]. The region with highly significant P-values contains several other genes besides PRKAG3. The allele effect of the most significant SNP (ALGA0087116) was 0.041 pH units (SE = 0.005), corresponding to 0.9 SD of the total additive genetic effect and approximately 20% of the additive genetic variance. Also, a single significant SNP (ALGA0106581) was observed 10 Mb from this region.
Because the SNPs at the PRKAG3 region had far stronger association with pH than the other SNPs on chromosome 15 or on other chromosomes, MARC0083357 was included as a covariate in the model, and GWAS was repeated. MARC0083357 was chosen because it was in complete LD with the most significant SNP ALGA87116, is located very close to PRKAG3 (62 kb from PRKAG3), and had a call rate of 1.0. The conditional GWAS revealed the same significant SNPs on chromosome 3 as the original GWAS. However, the significance of the SNPs in the first region on chromosome 15 (58.5 Mb–60.5 Mb) in the conditional analysis differed from the original GWAS depending on the LD between the SNPs and MARC0083357; e.g., the P-value for H3GA0044376 in the conditional GWAS was only 0.017 whereas the original P-value was 4.910E-07 (r 2 with MARC0083357 was 0.16). This reflects the long-distance LD found between MARC0083357 and several SNPs in the region 50 Mb–100 Mb (Fig. 3). However, some of the SNPs, e.g. ASGA0069650 (r 2 = 0.03), were segregating independently from MARC0083357, and the association with meat pH in the conditional GWAS for these SNPs was still relatively significant (Table 2). Additionally, several SNPs in the vicinity of PRKAG3 gave relatively small P-values, despite the fact that one of the most significant SNPs in that region was included in the model simultaneously (Table 2).

P-values (-log10-based) from the original and from the conditional association analysis and linkage disequilibrium with MARC0083357 measured as squared correlation (r2) for SNPs on chromosome 15
Haplotype analysis
Full LD was obtained for all significant SNPs and very strong LD (r 2 > 0.97) between all other SNPs in the region on chromosome 3, forming four haplotypes (Table 3). The association between the haplotypes (coded as 0: animal does not carry the haplotype, 1: animal carries one copy of the haplotype, and 2: animal carries two copies of the haplotype) and pH confirmed the positive effect of haplotype 1 on loin pH (Table 3). Compared to the rest of the haplotypes, the effect of haplotype 1 on loin pH was 0.018 (SE = 0.004), the same as in the single-SNP analysis.
The linkage disequilibrium structure (D’) of the first region on chromosome 15 (58.5 Mb–60.5 Mb) is presented in Fig. 4. The significant region from the original GWAS is approximately 2 Mb long and includes over 30 SNPs that passed the quality check. Even though the LD structure presented as D’ values (Fig. 4) is quite strong throughout the region, there are still 16 different haplotypes present. Haplotype 6, with very low frequency (0.05), showed strong association with loin pH: a haplotype effect of -0.036 pH units with SE = 0.008 and P-value = 6.6E-06 (Table 4). Also haplotype 7 showed a similar haplotype effect (-0.021, SE = 0.009) but with a higher P-value of 0.017. All other haplotypes gave a P-value > 0.05. Haplotypes 6 and 7 only have 30% of the alleles in common, but share three significant SNPs: H3GA0044376, ALGA0085452, and ASGA0069650 (see Table 2). Considering these two haplotypes together against all other haplotypes gave a P-value of 4.95E-07.

Linkage disequilibrium plots of the region 58.5 Mb–60.5 Mb on chromosome 15. SNPs presented in Table 2 are marked with bold lines
We also attempted a haplotype analysis for the second region of chromosome 15, but no further information was attained beyond the results presented for single SNPs. The reason for this is the length of the significant region, which created a very large number of possible haplotypes depending on the way haplotype blocks were defined.
Post-GWAS analysis
Based on the database annotation, a total of 56 genes were located next to the significant SNPs or regions identified in GWAS (Table 5). These genes were found to form main functional networks (Fig. 5). The genes on chromosome 3 (e.g. CREBBP and ADCY9) and on chromosome 15 (e.g. PRKAG3, IHH, WNT10A, STK36, and PLCD4 at 133.3 Mb–134.2 Mb) share statistically the most significant pathway, the Hedgehog signaling pathway. Other highlighted network pathways include Endocytosis, ErbB signaling, Faconiani anemia, RNA degradation, Non-homologous end-joining, Lysosome, and Primary bile acid biosynthesis.

Main functional group networks with pathway terms and genes as nodes. Red nodes represent pathways associated with genes on chromosome 3 (39.4 Mb–40.1 Mb); green and blue nodes are pathways associated with genes on chromosome 15 (58.5 Mb–60.5 Mb and 133.3 Mb–134.2 Mb, respectively). Pink and gray nodes are pathways shared between the regions. The most enriched terms per group are shown in bold according to enrichment significance from the ClueGO Cytoscape plug-in. Node size corresponds to enrichment significance. Nodes named in black are the observed genes
In addition, the four sets of genes in the significant chromosomal regions were used as input for the TFM-Explorer. Twenty-five TFs were identified through this analysis in each set. Based on the biological processes overrepresented in the BiNGO as well as on a literature review related to meat pH, we selected eight key TFs (Table 6) to construct a combined gene-TF network (Fig. 6). This network highlights the most connected genes within each set (e.g. TNS1, IGFBP5, VIL1, USP37, RQCD1, CRYBA2, PRKAG3, MAK16, NRG1, TRAP1, CLUAP1, and CREBBP) and provides an overview of shared TFs and genes across the single significant SNPs and regions on chromosomes 3 and 15.

Gene-transcription factor (TF) network. Transcription factors (pink nodes) and genes overlapping with relevant SNPs or regions for pH (yellow nodes are genes observed to be associated with single SNP group; red nodes are genes observed in chromosome 3 region (39.4 Mb–40.1 Mb); green nodes are genes observed in the first region on chromosome 15 (58.5 Mb–60.4 Mb); blue nodes are genes observed in the second region on chromosome 15 (133.3 Mb–134.2 Mb). The node size corresponds to the network analysis (Cytoscape) score, where bigger nodes represent higher edges density associated with the number of TF-binding sites. Purple square nodes are biological processes (GO terms) associated with muscle pH
Discussion
This article presents the results of GWAS and post-GWAS of pH measured from loin using the Finnish Yorkshire pig breed. A previous GWAS with 470 animals which were also included in the current study revealed two statistically significant (P-value < 2.0E-06) chromosomal regions: one on chromosome 2 and one on chromosome 15 (PRKAG3 region) [12]. As in the previous study, the region around PRKAG3 also gave the smallest P-value in this study. The SNPs on chromosome 2 found significant in the previous study [12] now reached a P-value of 5.77E-05 (ASGA0009838) that is still very close to significance and can be considered as “suggestive” finding. In the previous study, the best SNP on chromosome 3 (ASGA0014287) had a P-value of 5.89E-05. The best SNP on chromosome 15 at 58.5 Mb–60.5 Mb (ASGA0069650) also had an almost significant P-value (5.75E-06) in the previous study. Thus, increasing the number of observations from 470 to 703 changed the results somewhat. Results based on larger data are obviously more reliable than those based on smaller datasets.
Long-distance LD was observed between the SNPs in the PRKAG3 region and the other SNPs on chromosome 15. Including MARC0083357 in the model reduced the effect and significance of all SNPs on chromosome 15. Thus the significance observed in the first region on chromosome 15 may be “reflection” of the PRKAG3 region due to LD. However, the question why LD between these regions is so strong remains unsolved. Altogether, the mechanism on chromosome 15 that reduces pH in muscle post mortem is currently not yet fully understood. Beyond the original 200Q allele of PRKAG3, the functional variations could be due to other genes or to interaction between genes and transcription factors. Several studies have given evidence that I199V is highly associated with loin pH [32–35]. Also Rubio et al. [14] considered PRKAG3 as the best candidate in the region 132 Mb–135 Mb of chromosome 15 (the same region as detected in our study). However, based on a previous study by Uimari and Sironen [31], I199V is not directly associated with loin pH in the Finnish Yorkshire population; instead, the haplotype g.-157C–g.-58A–24E–199I is. Also Ryan et al. [36] reported a positive association of g.-157C with PRKAG3 expression and meat quality, and Zhang et al. [15] proposed that other genes besides PRKAG3 could be responsible for the association found in this region.
Post-GWAS
All the detected significant regions contain several genes. To find the most promising candidate genes within these regions and to understand the possible relationship between the candidate genes, a post-GWAS analysis was performed. Hedgehog signaling was the most significantly enriched pathway in the main functional group network (Fig. 5) comprising seven genes (PRKAG3, ADCY9, CREBBP, PLCD4, IHH, WNT10A, and STK36). The hedgehog signaling pathway has been cited as a key factor in the regulation of human adult tissue homeostasis and repair. It acts via multiple different routes to regulate distinct cellular outcomes, including the maintenance of plasticity [37]. Among the genes present in this pathway, ADCY9 is a type 9 adenylyl cyclase, which is involved in the main pathways promoting muscle relaxation by a cAMP (cyclic AMP) messenger [38]. PLCD4 is connected in the network through the calcium signaling and thyroid hormone signaling pathways. The phospholipase C enzyme promotes smooth muscle relaxation [39] and generates a second messenger IP3 that controls many cellular processes by inducing intracellular calcium mobilization [40].
The significant region of chromosome 3 contains two genes (CREBBP and ADCY9) involved in cyclic cAMP signaling. cAMP signaling mediates the effects of metabolism-controlling hormones, such as glucagon and epinephrine, and regulates energy homeostasis in multiple tissues [41]. cAMP is generated from ATP by adenylate cyclase enzymes, one of which is encoded by ADCY9 (adenylate cyclase 9) [42]. ADCY9 displays high expression in skeletal muscle and responds to beta-adrenergic receptor activation [43], which modulates Ca2+ release from the sarcoplasmic reticulum. Ca2+ release is a major contributor to pork meat quality, as is the case in the mutation R615C found in the pale, soft, and exudative (PSE) meat-related ryanodine receptor gene RYR1 [6]. cAMP activates protein kinase A (PKA), which controls metabolism either directly or through gene expression by phosphorylating the transcription factor CREB (cAMP-response element-binding protein), and consequently leading to the recruitment of its cofactor CBP (CREB binding protein) encoded by CREBBP [44]. Together, CREB and CBP regulate a plethora of metabolic target genes involved in glucose metabolism [41], which can potentially impact meat acidification. In addition to transcriptional regulation, PKA-mediated phosphorylation activates phosphorylase kinase, which stimulates glycogen phosphorylase to active glycogen release (glycogenolysis), which, in turn, is directly reflected on lactate production [41]. Moreover, PKA phosphorylates RYR1 and consequently regulates calcium release in skeletal muscle [45], providing another putative mechanism for the impact of ADCY9 on meat quality.
Each of the four sets of genes was also used to explore the promoter regions for enriched TFs, of which we selected the most relevant ones for meat pH to generate gene-TF networks. These networks were merged, enabling the visualization of the genes and the TFs that are common between genes in the different regions. The most connected TF (linked with a high number of genes) in this network is HNF4A, followed by PPARG and Nkx2-5. HNF4A encodes to hepatocyte nuclear factor 4α, which is essential to control the basal expression of genes involved in lipid metabolism and is indispensable for maintaining normal lipid homeostasis [46]. PPARG is the most studied isoform of the nuclear receptor superfamily, and performs an important role in regulating lipid and glucose homeostasis, in adipocytes differentiation, and in fatty acid storage [47]. Moreover, the polymorphism of this gene has been associated with meat quality traits in cattle [48]. The third most connected TF is Nkx2-5, which is a homeobox transcription factor known to be required for homeostasis of cardiac myocytes [49]. Briggs et al. [50] observed that Nkx2-5 knockout mice presented a reduced expression of ryanodine receptor 2 (RYR2), through which calcium is released from the sarcoplasmic reticulum. The corresponding skeletal muscle-related ryanodine receptor gene is RYR1. The most relevant biological processes of these TFs that affect meat pH are the homeostatic process (HNF4A and PPARG) and the regulation of calcium ion transport via voltage-gated calcium channel activity (Nkx2-5).
In the gene-TF network, HNF4A and PPARG are mainly connected with the genes on chromosome 3 and in the second region on chromosome 15, while genes identified in the first region of chromosome 15 are mainly connected with Nkx2-5. In the significant region on chromosome 3, TRAP1 is the most connected gene in the gene-TF network. TRAP1 encodes a mitochondrial chaperone protein that interacts with a calcium-binding protein, sorcin, and is thus involved in intracellular calcium concentration [51] and might also affect muscle pH. Of the genes in the first significant region on chromosome 15, the most connected gene in the gene-TF network is LOC102164569/NRG1 (pro-neuregulin-1, membrane-bound isoform-like). This gene is a member of the neuregulin growth factor gene family that is involved in the differentiation of embryonic muscle cells [52]. It has also been cited to be related to alterations of intracellular calcium homeostasis in humans [53] and to the marbling trait in Korean Hanwoo cattle [54]. We observed other genes besides PRKAG3 in the second region on chromosome 15 that are highlighted in the gene-TF network, such as VIL1, USP37, RQCD1, and CRYBA2. Among them, VIL1 has also been associated with meat pH and color in crossbred commercial pigs [15]. The most connected gene a bit further apart (700 kb) from PRKAG3 is TNS1 (tensin 1), which belongs to a focal adhesion gene family and interacts with actin filaments [55]. Recently, TNS1 has been identified as a candidate gene in GWAS for meat pH in Chinese Laiwu pigs [56]. Another gene, IGFBP-5, 2 Mb from PRKAG3, is a member of the IGF gene families. It has been suggested to be associated with meat quality, especially with pH in pigs [57]. Moreover, the mRNA level of this gene in muscle samples was observed to be significantly lower in Duroc pigs compared to other commercial breeds [58]. TNS1 and IGFBP5 are the most connected genes in the whole gene-TF network, and might thus be considered very stronger candidate genes for meat pH.
Conclusions
Three regions, one on chromosome 3 (39.4 Mb–40.1 Mb) and two on chromosome 15 (58.5 Mb–60.5 Mb and 133.3 Mb–134.2 Mb), were found to be highly associated with meat pH in Finnish Yorkshire. Additionally, several other SNPs up to a distance of 3 Mb from the known meat quality gene PRKAG3 proved significant. The significant regions harbored several genes, including two genes involved in cAMP signaling: ADCY9 and CREBBP. Based on post-GWAS analysis using functional and TF-gene networks, the most promising candidate genes for meat pH are ADCY9, CREBBP, TRAP1, NRG1, PRKAG3, VIL1, TNS1, and IGFBP5. The key TFs related to these genes are HNF4A, PPARG, and Nkx2-5. To conclude, we succeeded in identifying several genes which, based on SNP association, pathway, and transcription factor analysis, have potential to control muscle cell homeostasis and meat quality. However, functional studies are still needed to warrant the role of each of these genes in pork meat quality.
Abbreviations
- cAMP:
-
Cyclic AMP
- CBP:
-
CREB binding protein
- CR:
-
Call rate
- CREB:
-
cAMP-response element-binding protein
- dEBV:
-
Deregressed EBV
- EBV:
-
Estimated breeding value
- GO:
-
Gene ontology
- GWAS:
-
Genome-wide association study
- LD:
-
Linkage disequilibrium
- MAF:
-
Minor allele frequency
- Ne :
-
Effective population size
- PKA:
-
Protein kinase A
- PSE:
-
Pale, soft, and exudative
- REML:
-
Restricted maximum likelihood
- TF:
-
Transcription factor
- TFBS:
-
Transcription factor binding site
References
Bendall JR. Postmortem changes in muscle. In: Bourne GH, editor. The Structure and Function of Muscle. Vol. II Part 1. 2nd ed. New York.: Academic; 1973. p. 243–309.
Milan D, Jeon JT, Looft C, Amarger V, Robic A, Thelander M, Rogel-Gaillard C, Paul S, Iannuccelli N, Rask L, Ronne H, Lundström K, Reinsch N, Gellin J, Kalm E, Le Roy P, Chardon P, Andersson L. A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science. 2000;288(5469):1248–51.
Monin G, Sellier P. Pork of low technological quality with a normal rate of muscle pH fall in the immediate post-mortem period: The case of the Hampshire breed. Meat Sci. 1985;13(1):49–63.
Le Roy P, Naveau J, Elsen JM, Sellier P. Evidence for a new major gene influencing meat quality in pigs. Genet Res. 1990;55(1):33–40.
Hedegaard J, Horn P, Lametsch R, Møller HS, Roepstorff P, Bendixen C, Bendixen E. UDP-glucose pyrophosphorylase is upregulated in carriers of the porcine RN- mutation in the AMP-activated protein kinase. Proteomics. 2004;4(8):2448–54.
Fujii J, Otsu K, Zorzato F, De Leon S, Khanna VK, Weiler JE, O’Brien PJ, MacLennan DH. Identification of a mutation in porcine ryanodine receptor associated with malignant hyperthermia. Science. 1991;253(5018):448–51.
Ciobanu DC, Bastiaansen JWM, Lonergan SM, Thomsen H, Dekkers JCM, Plastow GS, Rothschild MF. New alleles in calpastatin gene are associated with meat quality traits in pigs. J Anim Sci. 2004;82(10):2829–39.
Ma J, Yang J, Zhou L, Ren J, Liu X, Zhang H, Yang B, Zhang Z, Ma H, Xie X, Xing Y, Guo Y, Huang L. A Splice Mutation in the PHKG1 gene causes high glycogen content and low meat quality in pig skeletal muscle. PLoS Genet. 2014;10(10), e1004710.
Becker D, Wimmers K, Luther H, Hofer A, Leeb T. A Genome-Wide Association Study to Detect QTL for Commercially Important Traits in Swiss Large White Boars. PLoS One. 2013;8(2), e55951.
Ma J, Yang J, Zhou L, Zhang Z, Ma H, Xie X, Zhang F, Xiong X, Cui L, Yang H, Liu X, Duan Y, Xiao S, Ai H, Ren J, Huang L. Genome-Wide Association Study of Meat Quality Traits in a White Duroc x Erhualian F2 Intercross and Chinese Sutai Pigs. PLoS One. 2013;8(5), e64047.
Nonneman DJ, Shackelford SD, King DA, Wheeler TL, Wiedmann RT, Snelling WM, Rohrer GA. Genome-wide association of meat quality traits and tenderness in swine. J Anim Sci. 2013;91(9):4043–50.
Uimari P, Sironen A, Sevón-Aimonen ML. Evidence for three highly significant QTL for meat quality traits in the Finnish Yorkshire pig breed. J Anim Sci. 2013;91(12):2001–11.
Sanchez MP, Tribout T, Iannuccelli N, Bouffaud M, Servin B, Tenghe A, Dehais P, Muller N, Del Schneider MP, Mercat MJ, Rogel-Gaillard C, Milan D, Bidanel JP, Gilbert H. A genome-wide association study of production traits in a commercial population of Large White pigs: Evidence of haplotypes affecting meat quality. Genet Sel Evol. 2014;46(1):12.
Rubio BYL, Gualdrón Duarte JL, Bates RO, Ernst CW, Nonneman D, Rohrer GA, King DA, Shackelford SD, Wheeler TL, Cantet RJC, Steibel JP. Implementing meta-analysis from genome-wide association studies for pork quality traits. J Anim Sci. 2015;93(12):5607–17.
Zhang C, Wang Z, Bruce H, Kemp RA, Charagu P, Miar Y, Yang T, Plastow T. Genome-wide association studies (GWAS) identify a QTL close to PRKAG3 affecting meat pH and colour in crossbred commercial pigs. BMC Genet. 2015;16(1):33.
Uimari P, Tapio M. Extent of linkage disequilibrium and effective population size in Finnish Landrace and Finnish Yorkshire pig breeds. J Anim Sci. 2011;89(3):609–14.
Verardo LL, Silva FF, Lopes MS, Madsen O, Bastiaansen JWM, Knol EF, Kelly M, Varona L, Lopes PS, Guimarães SEF. Revealing new candidate genes for reproductive traits in pigs: combining Bayesian GWAS and functional pathways. Genet Sel Evol. 2016;48:9.
Verardo LL, Lopes MS, Wijga S, Madsen O, Silva FF, Groenen MAM, Knol EF, Lopes PS, Guimarães SEF. After genome-wide association studies: Gene networks elucidating candidate genes divergences for number of teats across two pig populations. J Anim Sci. 2016;94(4):1446–58.
Sevón-Aimonen ML, Honkavaara M, Serenius T, Mäki-Tanila A, Puonti M. Genetic variation of loin and ham quality in Finnish Landrace and Large White pigs. Agric Food Sci. 2007;16(2):89–102.
Garrick DJ, Taylor JF, Fernando RL. Deregressing estimated breeding values and weighting information for genomic regression analyses. Genet Sel Evol. 2009;41:55.
Madsen P, Sørensen P, Su G, Damgaard LH, Thomsen H, Labouriau R. DMU – A Package for Analyzing Multivariate Mixed Models. 8th World Congress on Genetics Applied to Livestock Production. 2006.
Uimari P, Sironen A, Sevón-Aimonen ML. Whole-genome SNP association analysis of reproduction traits in the Finnish Landrace pig breed. Genet Sel Evol. 2011;43:42.
Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68(4):978–89.
Barrett JC, Fry B, Maller J, Daly MJ. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–5.
Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pagès F, Trajanoski Z, Galon J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25(8):1091–3.
Sandelin A, Alkema W, Engström P, Wasserman WW, Lenhard B. JASPAR: An open-access database for eukaryotic transcription factor binding profiles. Nucl Acids Res. 2004;32 suppl 1:D91–4.
Touzet H, Varré JS. Efficient and accurate P-value computation for position weight matrices. Algorithm Mol Biol. 2007;2(1):15.
Defrance M, Touzet H. Predicting transcription factor binding sites using local over-representation and comparative genomics. BMC Bioinforma. 2006;7:396.
Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics. 2005;21(16):3448–9.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003;13(11):2498–504.
Uimari P, Sironen A. A combination of two variants in PRKAG3 is needed for a positive effect on meat quality in pigs. BMC Genet. 2014;15:29.
Ciobanu D, Bastiaansen J, Malek M, Helm J, Woollard J, Plastow G, Rothschild M. Evidence for new alleles in the protein kinase adenosine monophosphate-activated gamma(3)-subunit gene associated with low glycogen content in pig skeletal muscle and improved meat quality. Genetics. 2001;159(3):1151–62.
Lindahl G, Enfält AC, von Seth G, Josell Å, Hedebro-Velander I, Andersen HJ, Braunschweig M, Andersson L, Lundström K. A second mutant allele (V199I) at the PRKAG3 (RN) locus-- I. Effect on technological meat quality of pork loin. Meat Sci. 2004;66(3):609–19.
Cherel P, Pires J, Glénisson J, Milan D, Iannuccelli N, Hérault F, Damon M, Le Roy P. Joint analysis of quantitative trait loci and major-effect causative mutations affecting meat quality and carcass composition traits in pigs. BMC Genet. 2011;12:76.
Rohrer GA, Nonneman DJ, Miller RK, Zerby H, Moeller SJ. Association of single nucleotide polymorphism (SNP) markers in candidate genes and QTL regions with pork quality traits in commercial pigs. Meat Sci. 2012;92(4):511–8.
Ryan MT, Hamill RM, O’Halloran AM, Davey GC, McBryan J, Mullen AM, McGee C, Gispert M, Southwood OI, Sweeney T. SNP variation in the promoter of the PRKAG3 gene and association with meat quality traits in pig. BMC Genet. 2012;13:66.
Petrova R, Joyner AL. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development. 2014;141(18):3445–57.
Kuo IY, Ehrlich BE. Signaling in muscle contraction. Cold Spring Harb Perspect Biol. 2015;7(2):a006023.
Calenda G, Suadicani SO, Iglesias R, Spray DC, Melman A, Davies KP. Silencing MaxiK activity in corporal smooth muscle cells initiates compensatory mechanisms to maintain calcium homeostasis. J Sex Med. 2011;8(8):2191–204.
Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361(6410):315–25.
Ravnskjaer K, Madiraju A, Montminy M. Role of the cAMP pathway in glucose and lipid metabolism. Handb Exp Pharmacol. 2016;233:29–49.
Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol. 2001;41:145–74.
Hacker BM, Tomlinson JE, Wayman GA, Sultana R, Chan G, Villacres E, Disteche C, Storm DR. Cloning, chromosomal mapping, and regulatory properties of the human type 9 adenylyl cyclase (ADCY9). Genomics. 1998;50(1):97–104.
Chrivia JC, Kwok RPS, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365(6449):855–9.
Reiken S, Lacampagne A, Zhou H, Kherani A, Lehnart SE, Ward C, Huang F, Gaburjakova M, Gaburjakova J, Rosemblit N, Warren MS, He KL, Yi GH, Wang J, Burkhoff D, Vassort G, Marks AR. PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: defective regulation in heart failure. J Cell Biol. 2003;160(6):919–28.
Yin L, Ma H, Ge X, Edwards PA, Zhang Y. Hepatic hepatocyte nuclear factor 4α is essential for maintaining triglyceride and cholesterol homeostasis. Arterioscler Thromb Vasc Biol. 2011;31(2):328–36.
Meirhaeghe A, Fajas L, Helbecque N, Cottel D, Auwerx J, Deeb SS, Amouyel P. Impact of the Peroxisome Proliferator Activated Receptor gamma2 Pro12Ala polymorphism on adiposity, lipids and non-insulin-dependent diabetes mellitus. Int J Obes Relat Metab Disord. 2000;24(2):195–9.
Fan YY, Zan LS, Fu CZ, Tian WO, Wang HB, Liu YY, Xin YP. Three novel SNPs in the coding region of PPARγ gene and their associations with meat quality traits in cattle. Mol Biol Rep. 2011;38(1):131–7.
Toko H, Zhu W, Takimoto E, Shiojima I, Hiroi Y, Zou Y, Oka T, Akazawa H, Mizukami M, Sakamoto M, Terasaki F, Kitaura Y, Takano H, Nagai T, Nagai R, Komuro I. Csx/Nkx2-5 is required for homeostasis and survival of cardiac myocytes in the adult heart. J Biol Chem. 2002;277:24735–43.
Briggs LE, Takeda M, Cuadra AE, Wakimoto H, Marks MH, Walker AJ, Seki T, Oh SP, Lu JT, Sumners C, Raizada MK, Horikoshi N, Weinberg EO, Yasui K, Ikeda Y, Chien KR, Kasahara H. Perinatal loss of Nkx2-5 results in rapid conduction and contraction defects. Circ Res. 2008;103(6):580–90.
Landriscina M, Laudiero G, Maddalena F, Amoroso MR, Piscazzi A, Cozzolino F, Monti M, Garbi C, Fersini A, Pucci P, Esposito F. Mitochondrial chaperone trap1 and the calcium binding protein sorcin interact and protect cells against apoptosis induced by antiblastic agents. Cancer Res. 2010;70(16):6577–86.
Kim D, Chi S, Lee KH, Rhee S, Kwon YK, Chung CH, Kwon H, Kang MS. Neuregulin stimulates myogenic differentiation in an autocrine manner. J Biol Chem. 1999;274(22):15395–400.
Giegling I, Genius J, Benninghoff J, Rujescu D. Genetic findings in schizophrenia patients related to alterations in the intracellular Ca-homeostasis. Prog Neuro-Psychopharmacol Biol Psychiatry. 2010;34(8):1375–80.
Lim D, Lee SH, Kim NK, Cho YM, Chai HH, Seong HH, Kim H. Gene co-expression analysis to characterize genes related to marbling trait in Hanwoo (Korean) cattle. Asian-Aust J Anim Sci. 2013;26(1):19–29.
Lo SH, Janmey PA, Hartwig JH, Chen LB. Interactions of tensin with actin and identification of its three distinct actin-binding domains. J Cell Biol. 1994;125(5):1067–75.
Xiong X, Liu X, Zhou L, Yang J, Yang B, Ma H, Xie X, Huang Y, Fang S, Xiao S, Ren J, Chen C, Ma J, Huang L. Genome-wide association analysis reveals genetic loci and candidate genes for meat quality traits in Chinese Laiwu pigs. Mamm Genome. 2015;26(3–4):181–90.
Wang W, Ouyang K, Shangguan X, Xu M. Association of Porcine IGF Binding Protein-5 Gene with Meat Quality. Biochem Genet. 2010;48(3):257–65.
Rejduch B, Oczkowicz M, Piestrzyńska-Kajtoch A, Piórkowska K, Witoń M, Rogoz M, Różycki M. Expression of IGFBP-3 and IGFBP-5 genes in muscles of pigs representing five different breeds. J Anim Feed Sci. 2010;19(4):554–63.
Miano JM, Long X, Fujiwara K. Serum response factor: Master regulator of the actin cytoskeleton and contractile apparatus. Am J Physiol Cell Physiol. 2007;292(1):C70–81.
Cui Y, Huang L, Elefteriou F, Yang G, Shelton JM, Giles JE, Oz OK, Pourbahrami T, Lu CYH, Richardson JA, Karsenty G, Li C. Essential Role of STAT3 in Body Weight and Glucose Homeostasis. Mol Cell Biol. 2004;24(1):258–69.
Wolff M, Jelkmann W, Dunst J, Depping R. The Aryl Hydrocarbon Receptor Nuclear Translocator (ARNT/HIF-1ß) is influenced by hypoxia and hypoxia-mimetics. Cell Physiol Biochem. 2013;32(4):849–58.
Kostanian IA, Vonarshenko AV, Lipkin VM. STAT1: a many-sided transcription factor. Bioorg Khim. 2010;36(1):15–28.
Acknowledgements
The authors wish to thank the Natural Resources Institute Finland (LUKE) for providing the genotypes for this study.
Funding
The research was funded by Suomen Sianjalostuksen Säätiö.
Availability of data and material
The datasets analyzed during the current study are not publicly available due to their possible commercial nature, but are available from the corresponding author upon reasonable request.
Authors’ contributions
LV and PU carried out the main data analysis, interpreted the results, and drafted the paper. VH interpreted the results and drafter the paper. MLSA and TS calculated the estimated breeding values. All authors read and approved the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval
The animal material included previously collected hair and semen samples; thus, no ethical approval was required. The authors had the pig owners’ approval to use the samples and to conduct this study
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Verardo, L.L., Sevón-Aimonen, ML., Serenius, T. et al. Whole-genome association analysis of pork meat pH revealed three significant regions and several potential genes in Finnish Yorkshire pigs. BMC Genet 18, 13 (2017). https://doi.org/10.1186/s12863-017-0482-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-017-0482-x