
Abstract
Background
The white-backed planthopper (WBPH), Sogatella furcifera (Horváth) (Hemiptera, Delphacidae), is a migratory pest of rice in Asia. Shandong Province, in northern China, is located on the migration pathway of WBPH between southern and northeast China. The potential sources of WBPH in northern China are poorly understood. We studied the sources of WBPH in Shandong Province by determining the population genetic structure of WBPH in 18 sites distributed in Shandong and in six regions of the Greater Mekong Subregion (GMS). We used mitochondrial gene and single-nucleotide polymorphism (SNP) markers for analysis.
Results
All of the WBPH populations studied in the seven regions had low genetic diversity. Pairwise FST values based on mtDNA ranged from − 0.061 to 0.285, while FST based on SNP data ranged from − 0.007 to 0.009. These two molecular markers revealed that 4.40% (mtDNA) and 0.19% (SNP) genetic variation could be explained by the interpopulation variation, while the rest came from intrapopulation variation. The populations in the seven geographic regions comprised four hypothetical genetic clusters (K = 4) not associated with geographic location. Eighty-four of 129 individuals distributed across the given area were designated as recent migrants or of admixed ancestry. Although the substantial migration presented, a weak but significant correlation between genetic and geographic distances was found (r = 0.083, P = 0.004).
Conclusion
The Greater Mekong Subregion was the main genetic source of WBPH in Shandong, while other source populations may also exist. The genetic structure of WBPH is shaped by both migration and geographic barriers. These results help clarify the migration route and the source of WBPH in northern China.
Similar content being viewed by others
Background
The white-backed planthopper (WBPH), Sogatella furcifera (Horváth), often causes serious yield losses to rice in Asia [1]. This pest usually migrates from tropical and subtropical regions toward northern or northeast Asia in spring and summer. At the end of the growing season in autumn, their offspring migrate back to their southern overwintering sites [2]. In 1970s and 1980s, a national collaborative study on the migration of WBPH was conducted in China. It was found that the spring migrants of WPBH were from the Indochina Peninsula, and they migrated into southern China [3]. The WBPH that migrated to China continue to move toward northern China on prevailing winds [4, 5]. Based on trajectory analysis, the rice paddy fields in the Greater Mekong Subregion (GMS), including Laos, Thailand, Vietnam, and parts of Yunnan Province in China, are considered to be important overwintering sites [6, 7]. Molecular marker data have confirmed that the extensive gene flow of WBPH occurs between Yunnan Province and neighboring countries in the GMS [8, 9].
Shandong Province in China is a typical region for growing single and midseason rice. Rice growing areas in Shandong Province are important because they are on the migration path of WBPH between southern and northeast China. Trajectories analysis demonstrated that some WBPH individuals migrating into China can reach northeastern China by late June or early July. The WBPH can then move between the Shandong and Liaoning provinces (or the Korean Peninsula) in both directions [4, 5]. The genetic background and detailed information of the potential source of WBPH in Shandong remain unclear.
Previous studies on WBPH migration mainly depended on the trajectory analysis method [4, 10]. Recently, molecular markers have been used for genetic studies of WBPH. These have included mitochondrial DNA genes (mtCOI) and nuclear genes (microsatellites) [8, 11, 12]. High-throughput sequencing of 2b-restriction site-associated DNA (2b-RAD) is available, and this can be used to scan the entire genome and identify large numbers of single-nucleotide polymorphisms (SNPs) [13]. This can help precisely evaluate the population differentiation among various geographic populations [14] and can determine disturbances of population structure caused by immigration [15].
In this study, we used both mitochondrial DNA and genome SNP markers to analyze the genetic diversity and connectivity of WBPH populations in seven geographic regions in Shandong Province, China and the GMS, including Cambodia, Laos, Myanmar (Burma), Thailand, Vietnam, and Yunnan Province. We characterized the genetic differentiation among WBPH populations and identified the possible source populations of WBPH in Shandong Province.
Results
Genetic diversity
A total of 663 bases in the mtDNA COI genes from 133 individuals (deposited in GenBank under Accession Nos. MN718018–MN718150) were obtained. Of the 663 sites, 653 were conserved, and 10, including nine singleton polymorphic sites and one parsimonious informative site, were variable. Of the 133 samples, 11 haplotypes were identified (Fig. 1, Table 1), of which nine were unique haplotypes and two were shared between populations. These two shared haplotypes (H1 and H2) accounted for 93.2% of the total haplotype frequency. The WBPH populations showed moderately low haplotype diversity and low nucleotide diversity. The overall haplotype diversity and nucleotide diversity were 0.453 and 0.00077, respectively (Table 1).

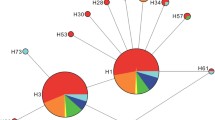
Haplotype network and haplotype frequencies of Sogatella furcifera obtained from sampling seven regions in China and Southeast Asian counties. a Haplotype network based on mtDNA COI. Circles representing haplotypes are proportional to the number of individuals per haplotype. Each hatch mark along a connecting line represents a change of one base pair. b Haplotype frequencies and distribution in seven regions. The color of each haplotype was the same as the network. ArcGIS desktop (version 10.0, https://www.esri.com/software/arcgis) was used to produce a distribution map based on the geographical coordinates of the localities. The base map for the depiction was obtained freely from the URL: https://www.naturalearthdata.com/downloads/
At SNP markers, all populations departed significantly from Hardy–Weinberg equilibrium due to a heterozygosity deficit (all P < 0.001). The observed heterozygosity ranged from 0.181 in Shandong (CN_SD) to 0.227 in Yunnan (CN_YN). The unbiased expected heterozygosity (uHE) values ranged from 0.227 in Myanmar (MM) to 0.272 in Shandong (CN_SD) (Table 2).
Population structure
Pairwise FST values computed from mtDNA data ranged from − 0.061 to 0.285, with an average FST of 0.098. Permutation tests showed that P value of FST were significant between CN_SD and Thailand population (TH) (P < 0.001), as well as between CN_YN and TH (P = 0.045) (Table 3). The haplotype network obviously displayed a two-star pattern with the common haplotype (H1 and H2) in the center of the two stars (Fig. 1). The FST values among populations represented in the Principal coordinate analysis (PCoA) showed that the TH was separated from the other populations (Fig. 2a).

Principal coordinates analysis (PCoA) based on pairwise FST values for mitochondrial DNA data (a) and for single-nucleotide polymorphism data (b) of Sogatella fucifera. PCoA is used to display genetic divergence among the populations. Population codes are given in Fig. 1
Pairwise FST values computed over SNP loci were quite low, ranging from − 0.007 to 0.009, with an average FST of 0.002. Pairwise FST value (0.009) between CN_SD and CN_YN was significant (P < 0.05) (Table 3). The PCoA result showed that 100% of the variation was explained by the first two axes. The first axis of the PCoA separated CN_SD and CN_YN (Fig. 2b).
The results of the AMOVA test on mtDNA and SNP markers in different populations are shown in Table 3. The global AMOVA of the data for the two molecular markers revealed that 4.40% (mtDNA) and 0.19% (SNP) genetic variation could be explained by the variation among populations, whereas the remainder came from variation within populations. Because of the low sample size in the Thailand population (TH), we reanalyzed global AMOVA excluding TH, and found no significant variation among the remaining populations. Based on the results of pairwise FST, we set the group = 2, which considered CN_SD as one group and the other populations as another group. Significant variability was found among SNP between these two groups (0.96%, P < 0.05) (Table 4).
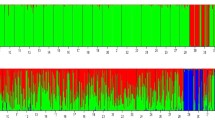
We also analyzed the population genetic structure based on SNP data using STRUCTURE software. The STRUCTURE analyses suggested that WBPH most likely forms four genetic clusters (Fig. 3). Indeed, for K = 4, the log-likelihood of the multilocus genotypic data was maximal and had low variance (Additional file 1: Figure S1). These clusters were not dependent on geographic regions because each population had the four genetic clusters, indicating a high level of gene flow.

STRUCTURE result of Sogatella furcifera when the given number of genotypic groups is 2, 3 and 4, respectively. Each bar represents a single individual from the designated population. Each color represents the proportion (from 0 to 1) of membership with regard to each hypothetical genetic cluster. Population codes are given in Fig. 1
Population assignment and isolation by distance
Based on population assignment test of the SNP data, 84 individuals were identified as migrants, there were connections among the Shandong population and other populations, all of which were expected to be a possible source of the Shandong population (Table 5). There were also frequent migrations among the Yunnan population and Southeast Asian populations. It is plausible that a high dispersal rate exists in Yunnan and Southeast Asia areas. However, migrants mainly moved from Yunnan population to the Southeast Asia areas populations.
In our mtDNA sequence data set, no isolation by distance (IBD) effects were detected with the standardized pairwise FST (r = − 0.028, P = 0.085; Fig. 4a). In contrast, there was a weak but significant IBD effect across the seven geographic regions in the SNP data (r = 0.083, P = 0.004; Fig. 4b).

Genetic distance (FST/1 − FST) vs. geographic distance for pairwise population comparisons based on mtDNA (a) and single-nucleotide polymorphism data (b) of Sogatella fucifera
Discussion
Genetic diversity of WBPH
The analysis based on the mitochondrial gene (COI) showed that all of the WBPH populations have relatively low genetic diversity. Only two dominant COI haplotypes widely exist in all populations suggesting a high-level gene flow among them. Among the 11 COI haplotypes identified, four haplotypes, including the two dominant haplotypes, were also found by previous study [8]. The number of haplotypes was much lower than the previous study [8] which found more rare haplotypes; it may be due to the difference in sampling size. Our study results demonstrate that the haplotype diversity was lower in Shandong Province than that in the GMS, which may be a consequence of founder events during migration. This result is consistent with that of [16] who demonstrated that range expansion can reduce genetic diversity in a long-distance migratory species.
The genome SNP markers (2b-RAD) analysis showed that a heterozygosity deficit existed in all populations. This result may be explained by demographic expansion, and it is consistent with [9] who also found that WBPH had a heterozygosity deficit during expansion. These findings suggest that WBPH may have non-random mating and intense migration in the sampled populations. With regard to spatial genetic variations, geographic factors play a weak role in WBPH populations. The AMOVA result indicated that there is only 4.40% (mtDNA) and 0.19% (SNP) genetic variation when all of the samples were grouped based on the geographic criteria. These results confirmed previous findings that WBPH migrates between the counties in GMS and China [9].
Population genetic structure of WBPH
The mtDNA and SNP data revealed different patterns of genetic connectivity among the WBPH populations (Table 3, Fig. 2). The main differences between markers concerned the genetic structure within the populations in Thailand and Shandong, where mtDNA clearly differentiated the Thailand population from the other populations, while SNP data separated the Shandong population from Yunnan population (Fig. 2). Because mtDNA is sensitive to founder effects and small population size, the probable loss or gain of a mtDNA haplotype will be greater for small populations, and it is often used to indicate migration among populations [17]. Based on the results of mtDNA, Shandong had close connectivity with the GMS. Therefore, it seems that the populations in Shandong may have come from the GMS.
SNP data provided information about the genetic structure of WPBH populations. Genetic connectivity among the GMS populations and Shandong population was close. Most of migrants were from Yunnan population as showed by results of population assignment. Compared with the mtDNA data, the SNP data were more consistent with the IBD pattern, perhaps as a result of high information of SNPs. Besides, mtDNA is often unsuitable for detecting isolation by distance [18]. This finding is consistent with the results of [19] who determined the effects of geographic isolation on the genetic structure of WBPH populations in Asia using microsatellite markers. A possible explanation for this might be that WBPH appears to have a stepwise migration. For example, it migrates from Southeast Asian areas into southern China, and then the second or later generations continue to move northward. Both genetic drift and local adaptation may influence the genetic variations of WBPH. As a consequence, geographic barriers and migration probably acted together to shape the genetic structure of WBPH.
In this study, the SNP markers showed that all of the populations exhibit high levels of admixture between the clusters identified with STRUCTURE. This indicates the occurrence of long distance migration events within geographical regions. Long-distance migration of WBPH allowed genetic mixing between populations from remote geographical origins [20]. This pattern may be common in other migratory insects, such as Helicoverpa spp. [21]. A population assignment test using the first-generation migrant detection method revealed Yunnan as the main source of WBPH in Shandong following by other areas in the GMS (Table 5). Our results also showed that the migrants moved from Yunnan to the Southeast Asian areas. Because of our sampling set, the sampling period in Yunnan (May) was two months later than in the Southeast Asian areas (March). It is impossible for the sampled Yunnan population move to the Southeast Asian areas. Therefore, Yunnan would probably supply migrants to the Southern Asian areas during autumn or winter. Regarding the bigger sample size in Yunnan than any other populations, we randomly sub-selected 20 individuals of Yunnan to repeat the assignment test. The result was much the same, although some details were different (Additional file 1: Table S1). The major migration routes of WBPH in East Asia were illustrated by [1] who reviewed previous studies of trajectory analyses. From mid-June to July, WBPH migrate from southern China to paddy fields in the middle and lower reaches of the Yangtze River, western Japan, and Korea. Because information on the migration of WBPH in Shandong is limited, many trajectory analysis and migration simulation usually neglect this area. Our results provide useful data for the migration route and source of WBPH in Shandong, which provide a better understanding of its migration routes. We inferred that WBPH in Shandong represents an important migration station. In this area, WBPH can establish connections with populations in Liaoning (North China) and Korea.
Dispersal of WBPH in China
Weather conditions are thought to expedite long-distance immigration of planthoppers [22]. In southern China, early season rice is planted in late March or early April. WBPH migrates into this area from Southeast Asian countries, such as Vietnam, Laos, and Thailand [3]. From May to June, WBPHs continually migrate into the Yangtzi River Valley. However, during this period, the emigration of WBPH in southern China is often hindered by heavy precipitation in southern China [23]. Because the migration of WBPH mainly depends on seasonal weather systems, WBPHs cannot migrate further north before mid-June [23]. Based on these data, we infer that the northward migration of WBPH during June and July largely contributes to the populations in the Shandong area.
Conclusion
This study demonstrated that WBPH populations have a low level of genetic diversity and a mixed genetic structure. We arranged the samples in chronological order, which depended on the occurrence of WBPH. Rice planting in Shandong mainly begins in May, and the WBPHs often have population outbreaks in July and August. Although the GMS were revealed as the main genetic source of WBPH in Shandong, WBPHs expand their range in a stepwise manner. Populations reproducing in other areas of China, such as Yangtze River Valley, Guangxi Zhuang Autonomous region, and Guizhou Province, may also be important sources. Future study is needed to examine more geographic populations and understand the temporal and spatial genetic structure of WBPH in China.
Methods
Insect samples
WBPH individuals were sampled in various geographic regions (China and Southeast Asia) (Table 6, Fig. 1). These comprised six sites in Yunnan Province; four sites in Shandong Province; two sites in Laos, Cambodia, and Vietnam, respectively; and one site in Myanmar and Thailand, respectively (Table 6). The sample size ranged from 7 to 43, with an average of 19. Samples were put in 95% ethanol and stored at − 20 °C until DNA extraction.
Mitochondrial COI sequencing
Insect DNA was extracted using the TIAMamp Micro DNA Kit (Tiangen, Beijing, China) according to the manufacturer protocol. The mtDNA COI gene was amplified using primers 2195-MF(5′-CTGGTTYTTTGGTCATCCRGARGT-3′) [24] and a newly designed reverse primer 2830-R(5′-CAATCAGCATAATCTGAATATCG-3′) (Sangon Biotech, Shanghai), which amplified a 635-bp fragment. The PCR reactions were performed in 20 μl solutions containing 1 × buffer, 0.32 mM of each dNTP, 1.0 mM of each primer, 1.0 unit of Taq DNA polymerase, and 2 μl of template DNA. PCR was performed under the following conditions: initial denaturation at 95 °C for 5 min, followed by 35 cycles of 1 min at 94 °C for denaturation, 1 min at 54 °C, for annealing and 1 min at 72 °C for elongation, and final extension at 72 °C for 5 min. The PCR products were electrophoresed in a 1.0% agarose gel in TAE and were sequenced bi-directionally. Sequencing quality was evaluated, and sequencing results were manually corrected using BioEdit 7.2.6 software [25], followed by BLAST for homology comparison in NCBI. The alignment of sequences was performed using multiple sequences of the Clustal W algorithm in MEGA7.0 [26].
2b-RAD sequencing and genotyping
The 2b-RAD sequencing and genotyping were outsourced to Shanghai OE Biotech Ltd. (Shanghai, China). Libraries were constructed following the 2b-RAD protocol [13]. Briefly, library preparation began with digestion of DNA samples. The BsaXI restriction enzyme (New England BioLabs, Ipswich, MA, USA) was used to prepare RAD libraries. Next, library-specific adaptors and the digestion products were linked with T4 DNA ligase. Ligation products were amplified by PCR, and the target band was excised from a 2% agarose gel. Finally, the paired-end RAD tags were sequenced on the Illumina Hiseq Xten platform (Illumina, San Diego, CA, USA). Quality filtering was conducted as follows: raw reads were trimmed to remove adaptors, and the terminal 2-bp positions were discarded to eliminate artifacts that might have arisen by ligation. Ambiguous bases (N) or reads of low quality (> 10 bp with quality less than Q20) were removed. SNPs were determined, and genotypes were called using a maximum-likelihood statistical model implemented in the software Stacks v1.32 [27].
Genetic variation analysis based on mitochondrial data
Numbers and distribution of haplotypes, composition of haplotypes in each population, numbers of unique haplotypes, within-population mean number of pairwise differences, and nucleotide diversity were assessed using DnaSP v.5.10 [28].The statistical parsimony network (also known as the TCS network) of haplotypes was analyzed using Popart ver. 1.7 [29, 30].
Genetic variation analysis based on SNP data
The genotype data contained information for each locus and each individual. The primary SNP loci number was 13,565, which could genotype all 133 individuals. We used Plink version 1.07 [31] to filter SNPs for genetic analysis. SNPs were filtered to meet the following criteria: (a) SNPs that were included in at least 80% samples of a population, (b) SNPs with a minor allele frequency (MAF) higher than 0.05, and (c) loci with strong deviations from the Hardy–Weinberg equilibrium (HWE, P < 0.0001) were removed. We excluded four samples which were from Shandong population that had too many missing data from further analyses reducing our sample size to 129 individuals. The final filtered SNP dataset had 1,108 SNP loci and was used for all downstream analyses. The parameters for population genetic analyses, that is, percentage of polymorphic loci (%poly), Shannon's information index (I), observed heterozygosity (HO), unbiased expected heterozygosity (uHE), and fixation index (F), were estimated by using GenALEx 6.5 [32, 33]. Hardy–Weinberg equilibrium (HWE), heterozygosity excess and deficit were tested by GENEPOP version 4.2.1 [34].
Population structure
We evaluated population genetic structure using five different approaches: (i) measuring genetic differentiation (FST) among populations, (ii) Principle Coordinate Analysis (PCoA) (iii) hierarchical analyses of molecular variance (AMOVA), (iv) Bayesian model-based clustering, and (v) Isolation by distance (IBD).
For mtDNA data, the pairwise FST were calculated using Arlequin v.3.5.1.2 [35] and using the Tamura–Nei model [36]. For SNP data, the pairwise FST were calculated using GenALEx. Principal coordinates analysis (PCoA) was used to find and plot the major pattern within a genetic distance matrix dataset. The PCoA using GenALEx, performed on genetic distance, was used to display genetic divergence among the populations. To determine the proportion of genetic variation that could be attributed to differences between sampling sites, hierarchal analyses of molecular variance (AMOVAs) were performed. A hierarchical AMOVA was performed using Arlequin, with 1000 permutations. Populations were grouped corresponding to two major criteria, i.e., geographical area, and population genetic structure, to test genetic homogeneity in different hierarchies.
The Bayesian approach was used to determine genetically distinct groups (or clusters) using the program STRUCTURE v.2.3.1 [37,38,39,40]. We set the length of the Burnin period at 10,000 and number of MCMC Reps after Burniin was 20,000. We set the K value from 1 to 7 and for each K the number of iterations was 10. To estimate the group number, we used the online calculation developed by [41]. We examined the change in Ln P(D) using the deltaK approach [42]. Because the STRUCTURE software showed results of each ten replications in the case of K = n, we used CLUMPP to average these results [43]. All of the data were visualized through DISTRUCT v.1.1 [44].
To estimate the admixture between geographic populations, we used a Bayesian assignment method as implemented in Geneclass2 [45]. This analysis identifies putative first-generation migrants among populations. To calculate individual probabilities of assignment to each population, we used the Monte-Carlo resampling method [46] with 1000 simulated individuals at probability thresholds of α = 0.05. Isolation by distance (IBD) analysis was performed using Mantel tests (1000 permutations) in GenAlex to find the correlation between genetic and geographic distances.
Availability of data and materials
The datasets of SNP genotype and summary statistics file can be accessed via Dryad. https://doi.org/10.5061/dryad.kwh70rz1c.
Abbreviations
- 2b-RAD:
-
2B-restriction site-associated DNA
- SNP:
-
Single-nucleotide polymorphism
- COI:
-
Cytochrome c oxidase subunit I
- FST :
-
F-statistics describe the statistically expected level of heterozygosity in a population
- WBPH:
-
The white-backed planthopper
- GMS:
-
The Greater Mekong Subregion
- IBD:
-
Isolation by distance
References
Otuka A. Migration of rice planthoppers and their vectored re-emerging and novel rice viruses in East Asia. Front Microbiol. 2013;4:309.
Otuka A, Matsumura M, Watanabe T, Dinh TV. A migration analysis for rice planthoppers, Sogatella furcifera (Horváth) and Nilaparvata lugens (Stål) (Homoptera: Delphacidae), emigrating from northern Vietnam from April to May. Appl Entomol Zool. 2008;43(4):527–34.
National Coordinated Research Group for White Back Planthoppers. Studies on the migration of white back planthoppers (Sogatella furcifera Horvath). Sci Agric Sin. 1981;5:25–31.
Shen JH, Shang JM, Liu GJ. Management of the white-backed planthopper, Sogatella furcifera in China: a mini-review. Chin J Rice Sci. 2003;17:7–22.
Hu C, Fu X, Wu K. Seasonal migration of white-backed planthopper Sogatella furcifera Horváth (Hemiptera: Delphacidae) over the Bohai Sea in northern China. J Asia-Pac Entomol. 2017;20(4):1358–63.
Hu SJ, Fu DY, Han ZL, Ye H. Density, demography, and influential environmental factors on overwintering populations of Sogatella furcifera (Hemiptera: Delphacidae) in southern Yunnan, China. J Insect Sci. 2015;15:58.
Sun SS, Bao YX, Wu Y, Lu MH, Tuan HA. Analysis of the huge immigration of Sogatella furcifera (Hemiptera: Delphacidae) to Southern China in the spring of 2012. Environ Entomol. 2018;47(1):8–18.
Li XY, Chu D, Yin YQ, Zhao XQ, Chen AD, Khay S, Douangboupha B, Kyaw MM, Kongchuensin M, Ngo VV, et al. Possible source populations of the white-backed planthopper in the Greater Mekong subregion revealed by mitochondrial DNA analysis. Sci Rep. 2016;6(1):39167.
Yin Y, Li X, Chu D, Zhao X, Sathya K, Douangboupha B, Kyaw MM, Kongchuensin M, Somrith A, Ngo VV, et al. Extensive gene flow of white-backed planthopper in the Greater Mekong Subregion as revealed by microsatellite markers. Sci Rep. 2017;7(1):15905.
Riley JR, Smith AD, Reynolds DR. The feasibility of using vertical-looking radar to monitor the migration of brown planthopper and other insect pests of rice in China. Insect Sci. 2003;10(1):1–19.
Matsumoto Y, Matsumura M, Sanada-Morimura S, Hirai Y, Sato Y, Noda H. Mitochondrial cox sequences of Nilaparvata lugens and Sogatella furcifera (Hemiptera, Delphacidae): low specificity among Asian planthopper populations. Bull Entomol Res. 2013;103(4):382–92.
Mun JH, Song YH, Heong KL, Roderick GK. Genetic variation among Asian populations of rice planthoppers, Nilaparvata lugens and Sogatella furcifera (Hemiptera: Delphacidae): mitochondrial DNA sequences. Bull Entomol Res. 1999;89(3):245–53.
Wang S, Meyer E, McKay JK, Matz MV. 2b-RAD: a simple and flexible method for genome-wide genotyping. Nat Methods. 2012;9(8):808–10.
Morin PA, Luikart G, Wayne RK. the SNPwg: SNPs in ecology, evolution and conservation. Trends Ecol Evol. 2004;19(4):208–16.
Morin PA, Martien KK, Taylor BL. Assessing statistical power of SNPs for population structure and conservation studies. Mol Ecol Resour. 2009;9(1):66–73.
Pierce AA, Zalucki MP, Bangura M, Udawatta M, Kronforst MR, Altizer S, Haeger JF, de Roode JC. Serial founder effects and genetic differentiation during worldwide range expansion of monarch butterflies. Proc R Soc B. 2014;281(1797):20142230.
Roderick GK. Geographic structure of insect populations: gene flow, phylogeography, and their uses. Annu Rev Entomol. 1996;41(1):325–52.
Teske PR, Golla TR, Sandoval-Castillo J, Emami-Khoyi A, van der Lingen CD, von der Heyden S, Chiazzari B, Jansen van Vuuren B, Beheregaray LB. Mitochondrial DNA is unsuitable to test for isolation by distance. Sci Rep. 2018;8(1):8448.
Nam HY, Kim KS, Lee JH. Population genetic structure and putative migration pathway of Sogatella furcifera (Horváth) (Hemiptera, Delphacidae) in Asia. Bull Entomol Res. 2019;109(4):453–62.
Sun JT, Jiang XY, Wang MM, Hong XY. Development of microsatellite markers for, and a preliminary population genetic analysis of, the white-backed planthopper. Bull Entomol Res. 2014;104(6):765–73.
Jones CM, Parry H, Tay WT, Reynolds DR, Chapman JW. Movement ecology of pest Helicoverpa: implications for ongoing spread. Annu Rev Entomol. 2019;64(1):277–95.
Kisimoto R. Synoptic weather conditions inducing long-distance immigration of planthoppers, Sogatella furcifera Horvath and Nilaparvata lugens Stal. Ecol Entomol. 1976;1(2):95–109.
Chen H, Chang X-L, Wang Y-P, Lu M-H, Liu W-C, Zhai B-P, Hu G. The early northward migration of the white-backed planthopper (Sogatella furcifera) is often hindered by heavy precipitation in southern China during the preflood season in May and June. Insects. 2019;10(6):158.
Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am. 1994;87(6):651–701.
Hall T. BioEdit: a user-friendly biological sequence alignment program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–8.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4.
Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH. Stacks: building and genotyping loci de novo from short-read sequences. G3. 2011;1(3):171–82.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–2.
Clement M, Snell Q, Walker P, Posada D, Crandall K. TCS: estimating gene genealogies. In: Parallel and Distributed Processing Symposium, International Proceedings. 184; 2002.
Leigh JW, Bryant D. Popart: full-feature software for haplotype network construction. Methods Ecol Evol. 2015;6(9):1110–6.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75.
Peakall R, Smouse PE. GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes. 2006;6(1):288–95.
Peakall R, Smouse PE. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics. 2012;28:2537–9.
Rousset F. GENEPOP’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour. 2008;8(1):103–6.
Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10(3):564–7.
Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10(3):512–26.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–59.
Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164(4):1567–87.
Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes. 2007;7(4):574–8.
Hubisz MJ, Falush D, Stephens M, Pritchard JK. Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour. 2009;9(5):1322–32.
Earl DA, vonHoldt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2012;4(2):359–61.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14(8):2611–20.
Jakobsson M, Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23(14):1801–6.
Rosenberg NA. Distruct: a program for the graphical display of population structure. Mol Ecol Notes. 2004;4(1):137–8.
Piry S, Alapetite A, Cornuet JM, Paetkau D, Baudouin L, Estoup A. GENECLASS2: a software for genetic assignment and first-generation migrant detection. J Hered. 2004;95(6):536–9.
Paetkau D, Slade R, Burden M, Estoup A. Genetic assignment methods for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Mol Ecol. 2004;13(1):55–65.
Acknowledgements
Not applicable.
Funding
This research was supported by the Shandong Modern Agricultural Technology & Industry System (SDAIT-17-07), First Class Grassland Science Discipline Programme in Shandong Province of China, the National Natural Science Foundation of China (No.31860504), and the Key Research and Development Program of Yunnan Province (No.2019IB007). These funding sources played no role in the design of the study, the collection, analysis, and/or interpretation of data, and the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
DC designed the experiments; NY, AC, YY and XL collected the data; ZD performed the analysis; NY, ZD and DC wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
(A) Delta k value of data across 10 replicates of STRUCTURE, where k = 4 is shown as the best fit of the data for the highest level of hierarchical genetic structure. (B) The mean lnP(D|K) and SD for each k where the model of k = 2, 3 or 4 is indicated as the best fit. Table S1. Assignment test for Sogatella furcifera individuals in the seven geographic populations. Individuals are presented in rows according to their sampling locations as individuals assigned to their own population (self) and those assigned to other putative source population. Individuals in CN_YN were randomly selected as 20 individuals participated the analysis.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Yang, N., Dong, Z., Chen, A. et al. Migration of Sogatella furcifera between the Greater Mekong Subregion and northern China revealed by mtDNA and SNP. BMC Evol Biol 20, 154 (2020). https://doi.org/10.1186/s12862-020-01722-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-020-01722-4