
Abstract
Background
The Caribbean offers a unique opportunity to study evolutionary dynamics in insular mammals. However, the recent extinction of most Caribbean non-volant mammals has obstructed evolutionary studies, and poor DNA preservation associated with tropical environments means that very few ancient DNA sequences are available for extinct vertebrates known from the region’s Holocene subfossil record. The endemic Caribbean eulipotyphlan family Nesophontidae (“island-shrews”) became extinct ~ 500 years ago, and the taxonomic validity of many Nesophontes species and their wider evolutionary dynamics remain unclear. Here we use both morphometric and palaeogenomic methods to clarify the status and evolutionary history of Nesophontes species from Hispaniola, the second-largest Caribbean island.
Results
Principal component analysis of 65 Nesophontes mandibles from late Quaternary fossil sites across Hispaniola identified three non-overlapping morphometric clusters, providing statistical support for the existence of three size-differentiated Hispaniolan Nesophontes species. We were also able to extract and sequence ancient DNA from a ~ 750-year-old specimen of Nesophontes zamicrus, the smallest non-volant Caribbean mammal, including a whole-mitochondrial genome and partial nuclear genes. Nesophontes paramicrus (39-47 g) and N. zamicrus (~ 10 g) diverged recently during the Middle Pleistocene (mean estimated divergence = 0.699 Ma), comparable to the youngest species splits in Eulipotyphla and other mammal groups. Pairwise genetic distance values for N. paramicrus and N. zamicrus based on mitochondrial and nuclear genes are low, but fall within the range of comparative pairwise data for extant eulipotyphlan species-pairs.
Conclusions
Our combined morphometric and palaeogenomic analyses provide evidence for multiple co-occurring species and rapid body size evolution in Hispaniolan Nesophontes, in contrast to patterns of genetic and morphometric differentiation seen in Hispaniola’s extant non-volant land mammals. Different components of Hispaniola’s mammal fauna have therefore exhibited drastically different rates of morphological evolution. Morphological evolution in Nesophontes is also rapid compared to patterns across the Eulipotyphla, and our study provides an important new example of rapid body size change in a small-bodied insular vertebrate lineage. The Caribbean was a hotspot for evolutionary diversification as well as preserving ancient biodiversity, and studying the surviving representatives of its mammal fauna is insufficient to reveal the evolutionary patterns and processes that generated regional diversity.
Similar content being viewed by others

Background
Accurate reconstruction of past species richness in recently extinct faunas is essential, in order to understand evolutionary history and the magnitude of human impacts on biodiversity through time. It is therefore necessary to determine the extent to which variation in faunal samples from environmental archives represents species-level differentiation, as opposed to intraspecific variation within or between conspecific populations. This can require both genetic and/or morphometric approaches [1,2,3,4], as well as comparative understanding from extant taxa about levels of variation expected within related living populations [5, 6]. In particular, uncertainty about past biodiversity levels can limit understanding of the timing and dynamics of lineage diversification seen during adaptive radiations on island systems, which are widely regarded as “natural laboratories” for studying evolution [7,8,9]. Insular taxa frequently experience distinctive patterns of body size change [10], but the speed at which diversifying insular lineages are able to occupy novel niches through size change remains poorly understood [11]. However, islands have experienced disproportionately high levels of human-caused extinction during recent millennia, with many extinct taxa known only from morphologically incomplete and molecularly degraded specimens preserved in the recent fossil or zooarchaeological records, hindering investigation of taxonomic baselines and evolutionary patterns [12,13,14].
Islands have been of relatively limited value for investigating adaptive dynamics in mammals, a well-studied group that have otherwise provided extensive insights into evolutionary trends and mechanisms, because most non-volant mammal groups show limited ability to colonise island systems through overwater dispersal [13]. The insular Caribbean is an exception. This island region has acted as an important study system for investigating ecological drivers and evolutionary dynamics of morphological differentiation in novel environments for many animal groups [9, 15], and it was also colonised by several land mammal lineages, providing an important opportunity to understand mammalian evolutionary dynamics within an insular context. However, nearly all of the region’s endemic insular mammal fauna became extinct following multiple waves of human colonisation from the mid-Holocene onwards, which led to the loss of over 100 mammal species or distinct island populations [12, 14, 16]. There is continuing uncertainty over the taxonomic status of many extinct Caribbean mammals, with many recently recognised species now considered dubious or invalid [17, 18] and other unstudied populations potentially representing undescribed species [19, 20], but molecular studies have been limited by poor DNA preservation under high thermal ages represented by the Caribbean’s hot, humid tropical conditions [21,22,23].
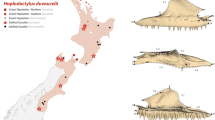
The islands of the western Caribbean (the Greater Antilles) contain a radiation of eulipotyphlan insectivores in the endemic suborder Solenodonota, including the highly threatened solenodons (Solenodontidae) of Cuba and Hispaniola (Atopogale cubana and Solenodon paradoxus) [22, 24], and the recently extinct Caribbean “island-shrews” (Nesophontidae), which comprised the single genus Nesophontes (Fig. 1). Nesophontids were the smallest endemic non-volant Caribbean land mammals, and probably became extinct around 500 years ago following European-era introduction of black rats (Rattus rattus) [25, 26]. Some islands (Puerto Rico, Cayman Brac, Grand Cayman) contained single Nesophontes species, but multiple species, distinguished through morphological and/or size differences, have been described from Cuba and Hispaniola [27,28,29,30]. The validity of most Cuban species is disputed, and nesophontid diversity from this island is uncertain [31], with some authorities suggesting that only a single species was actually present [29]. Size variation shown within Nesophontes samples from Puerto Rico has also been interpreted as probable intraspecific variation [32, 33].

a, Map of the Caribbean, showing former distribution of Nesophontes (red stars) and relative size differences between Hispaniolan Nesophontes species. Basemap source: Wikimedia Commons. b, Map of Hispaniola, showing locations of fossil sites containing Nesophontes specimens included in this study. Map generated using Adobe Illustrator CS6 (www.adobe.com). Key: 1, Trou Diable; 2, Trou Fon Pelen; 3, Trou Jean Paul; 4, Cerro de San Francisco; 5, Cueva los Tres Ojos; 6, Cueva de Bosque Humido
Three nesophontid species have been described from Hispaniola, the second-largest Caribbean island (divided politically into the Dominican Republic and Haiti), which are differentiated largely on the basis of size [28]: Nesophontes paramicrus (39-47 g), N. hypomicrus (21-24 g), and N. zamicrus (~ 10 g) (body mass estimates from ref. [14], calculated using predictive regression equations in ref. [34] based on molar measurements) (Fig. 1). All three species have been reported to co-occur in recent subfossil deposits across Hispaniola [25, 28, 35,36,37], suggesting they might represent a rare example of an insular mammalian adaptive radiation driven by niche availability for differing body size classes. However, the taxonomic validity and interrelationships of Hispaniolan nesophontids have not been investigated since their original description by Miller in 1929 [28], hindering understanding of past Caribbean mammalian diversity and the history and mechanism of evolutionary diversification in recently extinct endemic mammals.
Brace et al. [22] were able to recover ancient DNA (aDNA) from a Nesophontes paramicrus specimen from Cueva de Bosque Humido (= Cueva del Carpintero of ref. [38]; 19.077389 N, 69.477389 W), Los Haitises National Park, Hato Mayor Province, northern Dominican Republic (Fig. 1), allowing them to resolve phylogenetic relationships between the Solenodontidae and Nesophontidae. Additional Nesophontes specimens collected from the same rich surficial accumulation of small vertebrate skeletal elements in this cave match the morphological diagnoses of N. hypomicrus and N. zamicrus [28], with a direct date of 734 ± 24 yr BP available for a N. hypomicrus cranium from this accumulation [22]. In order to understand the taxonomic status, phylogenetic relationships and divergence timing of Hispaniola’s extinct eulipotyphlan fauna, we used morphometric analyses to quantify phenotypic variation in late Quaternary Nesophontes samples from across the island, and aDNA techniques to obtain genetic sequence data for multiple Nesophontes size morphs from the same late Holocene palaeontological site. Using this combined morphometric-genetic approach of across-island and within-landscape variation, we are able to reconstruct the past diversity and dynamics of evolutionary differentiation in an enigmatic group of extinct mammals, with wider implications for understanding the evolutionary processes that have generated mammalian diversity on islands.
Results
We measured 65 Nesophontes mandibles from six late Quaternary fossil sites across Hispaniola, including from Cueva de Bosque Humido and other sites in both the Dominican Republic and Haiti (Fig. 1; Tables S1-S2). Cluster analysis of linear morphometric PCAs identified three clusters (parameters of automatically selected best model based on BIC = ellipsoidal, equal shape and orientation) (Fig. 2). Measured specimens from Cueva de Bosque Humido are distributed across all three clusters. Specimen classification uncertainty was < 0.05 except for one specimen (Table S2). Only the first two principal component loadings, which represent 97.0% of variation explained by PCA, were needed to achieve a stable cluster model. PC1 was highly correlated (> 0.94) with all mandibular measurements. Reanalysis using log-transformed measurements resulted in identical classification of specimens to each cluster (results not shown). There was no overlap in any measurements between clusters. ANOVAs for all measurements showed statistically significant differences between clusters, and post-hoc Tukey tests revealed statistically significant differences between all three clusters for every measurement (all P ≤ 0.005).

Principal Component Analysis of linear measurement data for 65 Hispaniolan Nesophontes mandibles, where unsupervised cluster analysis of principal component loadings identifies three discrete clusters. Specimens highlighted with open circles are from Cueva de Bosque Humido
We were able to extract and sequence aDNA from two samples from Cueva de Bosque Humido that were referable on morphological criteria to the extinct endemic Hispaniolan insectivore Nesophontes zamicrus, although unfortunately we were unable to recover aDNA from available samples referable to N. hypomicrus from this site. The two N. zamicrus samples differed markedly in the number of reads that mapped to the N. paramicrus mitochondrial genome, and only the sample with the highest endogenous content was analysed further. We obtained a whole-mitochondrial genome (15,280 bp) and partial nuclear genes (APP, BMI1, CREM, PLCB4, ADORA3, APOB, ADRA2B, ADRAB2) for N. zamicrus.
Maximum likelihood and Bayesian analyses generated almost identical well-resolved topologies, with the exception of the weakly-supported placement of the talpid genera Galemys and Urotrichus (Fig. 3, Fig. S2). Our analyses strongly support Nesophontes paramicrus and N. zamicrus as a monophyletic clade (Figs. 3 and 4, Fig. S2). These Hispaniolan Nesophontes species diverged from each other relatively recently, with a mean estimated divergence date of 0.699 Ma during the Middle Pleistocene (95% HPD: 0.392–1.111 Ma). Pairwise genetic distance values for N. paramicrus and N. zamicrus based on mitochondrial and nuclear genes are all relatively low (RAG1, 0.1%; BRCA1, 0.4%; BDNF, 0.6%; CREM, 2.2%; cyt b, 2.2%; 12S, 5.3%), but these values are all within or above the ranges of comparative pairwise genetic distances estimated between other congeneric eulipotyphlan species for which GenBank data are available (RAG1, 0.1–3.6%; BRCA1, 0.2–8.8%; BDNF, 0.0–1.5%; CREM, 0.0–0.5%; cyt b, 0.3–15.6%; 12S, 2.2–9.5%) (Tables S7-S12).

Bayesian inference phylogeny, including Nesophontes paramicrus, N. zamicrus, extant eulipotyphlan species with available whole-mitochondrial genome data and nuclear genes (APP, BMI1, CREM, PLCB4, ADORA3, APOB, ADRA2B, ADRAB2), and inclusive of all out-group taxa. Node values indicate posterior probability. Scale indicates nucleotide substitutions per site

Time-calibrated phylogeny generated in BEAST using available whole-mitochondrial genome data and nuclear genes (APP, BMI1, CREM, PLCB4, ADORA3, APOB, ADRA2B, ADRAB2), showing estimated divergence dates for Nesophontes paramicrus and N. zamicrus. Estimates of median divergence dates shown above nodes in red. Node bars indicate 95% highest posterior density values. Posterior probability values shown on branches in grey
Discussion
Our combined morphometric-genetic framework provides a robust new baseline for interpreting morphological variation seen in subfossil samples of nesophontid island-shrews from the large Caribbean island of Hispaniola, and our complementary analyses clarify levels of both taxonomic diversity and evolutionary differentiation within this recently extinct island mammal fauna, providing a template for future investigation of biodiversity patterns observed in environmental archives. Nesophontid mandibular samples from late Quaternary fossil sites across Hispaniola fall into three non-overlapping clusters in morphometric analysis, with between-cluster differentiation on PC1 highly correlated with our measurement indices of specimen size. Several explanations have been proposed to account for variation in within-island nesophontid samples, including sexual size dimorphism and allochronic plasticity as well as taxonomic differentiation [32, 33]. Sexual dimorphism is minimal in the extant eulipotyphlan radiation [24], within which nesophontids are phylogenetically nested [22], making this explanation unlikely, and it would not be expected to exhibit a pattern of three-way clustering in PCA. Most late Quaternary sites across Hispaniola, including several sampled for this study, have not been dated, making potential allochronic plasticity difficult to evaluate. However, the subfossil deposit at Cueva de Bosque Humido is a surficial owl accumulation with multiple direct AMS dates from within the last thousand years [22], and measured nesophontid specimens from this site fall into all three morphometric clusters identified in our PCA, showing that this pattern of morphometric differentiation was present in the late Holocene and was seen across individuals from the same landscape (Fig. 2). These results therefore provide statistical support for the former co-occurrence of three size-differentiated Nesophontes species on Hispaniola, as proposed in the 1920s by Miller [28] but not subsequently investigated using rigorous quantitative methods.
Complementing our morphometric findings, our genetic data indicate that two nesophontid specimens from the same late Holocene deposit in Cueva de Bosque Humido, which correspond morphologically to different described Hispaniolan Nesophontes species (N. paramicrus and N. zamicrus), exhibit genetic differentiation (e.g., pairwise genetic distance values for cyt b = 2.2%) and have a mean estimated divergence date during the Middle Pleistocene (0.699 Ma). In comparison, genetic distances for cyt b within extant small mammal species (including eulipotyphlans, rodents and bats), at both within-population and between-population or between-subspecies levels, are typically < 2% and usually considerably lower within a single population [39,40,41,42]. Island-endemic taxa often have even lower genetic variation compared to mainland taxa, possibly associated with stronger effects of genetic drift in small, geographically restricted island populations [43,44,45,46], making the genetic differentiation observed in our study even more noteworthy. The level of genetic differentiation seen between ~ 750 year-old nesophontid specimens from the same landscape in northern Hispaniola is thus much greater than would be expected for within-population variation in a single species, and therefore provides further evidence that these individuals likely represented different species.
Comparison of our Nesophontes genetic divergence data with extant eulipotyphlan insectivores is limited by the general absence of well-resolved species-level molecular phylogenies for other eulipotyphlan radiations. Pairwise genetic distances for Hispaniolan Nesophontes fall within the overall range of available comparative data for extant eulipotyphlan species-pairs that are diagnosed as valid species on the basis of morphological criteria. While differences between the two nesophontids are low at mitochondrial loci, pairwise genetic distances between incomplete nuclear genes analysed in our study are more comparable to other species. The youngest reported species divergences within many other eulipotyphlans are much older in comparison than observed in Hispaniolan Nesophontes. The youngest reported divergences date from earlier in the Pleistocene in erinaceids (Erinaceus concolor–E. roumanicus, mean = 0.89 Ma, 95% HPD = 0.4–1.4 Ma; E. amurensis–E. europaeus, mean = 1.06 Ma, 95% HPD = 0.6–1.6 Ma) [47] and in talpids (Mogera imaizumi–M. tokudae; mean = 0.8–1.0 Ma across alternative phylogenetic models; 95% HPD across all models = 0.1–2.3 Ma) [48]. Reported intraspecific divergence events for both temperate and tropical talpids also date from the Early-Middle Pleistocene (1.4 to 0.17 Ma) [42, 49]. Conversely, the youngest reported species divergences in soricids are comparable to Hispaniolan Nesophontes. For example, the Anourosorex squamipes–A. yamashinai divergence is dated to 0.66 Ma (95% HPD = 0.18–1.35 Ma) [50], and divergence within the Crocidura suaveolens complex is dated to 1.9–0.9 Ma, with different lineages exhibiting distinct morphotypes that are likely to represent separate species [51]. Similarly, divergence of 11 tropical Sorex species from Mexico and Guatemala is estimated to have begun 8.91 Ma, but continued until as recently as the species-level split between S. cinereus and S. milleri at 0.68 Ma [52]. These comparative data are thus consistent with recognition of species-level status for Nesophontes paramicrus and N. zamicrus based on our genetic data.
Mitochondrial sequence data for over 70 mammalian sister-species pairs have a median divergence date of 3.2 Ma under a standard (2%/MY) molecular clock. However, after using median phylogroup separation times to correct for the fact that speciation occurs over time and is not a point event, at least 50% of these mammalian sister-species speciation events are calculated to have occurred during the Pleistocene [53]. Indeed, well-recognised species pairs in many other mammal groups also show similarly young divergences to Hispaniolan Nesophontes, and many of these groups have much longer generation lengths than eulipotyphlans, so these temporal divergence estimates reflect an even shorter number of generations. For example, polar bears (Ursus maritimus) and brown bears (U. arctos) are estimated to have diverged 0.34–0.48 Ma [54]. Recent and rapid speciation has also been demonstrated in Rattus, with multiple species arising in the Middle Pleistocene [55]. Other recent species-level divergences include many boreal species such as Eurasian and American lynx (Lynx lynx and L. canadensis) and three species of lemmings (Lemmus) [56]. These wider comparisons with our genomic data further support recognition of species-level status for Nesophontes paramicrus and N. zamicrus.
Many of these comparative examples of other mammal species exhibiting recent divergences are from mid- or high-latitude regions, where novel environmental conditions, geologically sudden barriers to gene flow, and population movement, fragmentation and expansion/contraction associated with tracking changing habitats during Late Quaternary glacial-interglacial cycles could have promoted rapid evolutionary differentiation. In contrast, population-level divergences often date further back to the Early Pleistocene or Pliocene in many tropical environments [57]. However, Late Quaternary fluctuating climates and environmental change are known to have caused considerable disruption to Hispaniola’s ecosystems during the estimated period of divergence between Nesophontes paramicrus and N. zamicrus, including marine inundation of low-lying valleys that generated transient barriers to gene flow [58, 59], and habitat shifts between mesic and xeric conditions [60]. A shift in glacial-interglacial cyclicity around 0.8 Ma, just before the mean estimated divergence date for Hispaniolan Nesophontes, could have exacerbated population-level effects of these environmental changes [61]. Such geologically recent processes could potentially have driven population fragmentation and speciation in Hispaniolan Nesophontes.
Hispaniola’s two extant endemic non-volant land mammals, the Hispaniolan solenodon and Hispaniolan hutia (Plagiodontia aedium), show similar between-population pairwise genetic distances (cyt b: solenodon, 1.1–2.2%; hutia, 1.03–3.2%) and divergence date estimates (means = 0.436–0.594 Ma for divergences between three hutia populations) to Hispaniolan Nesophontes species [62, 63]. However, these taxa instead exhibit low alpha diversity and high beta (between-landscape) diversity, with separate lineages distributed allopatrically rather than sympatrically across Hispaniola’s geologically and environmentally heterogeneous landscapes. Allopatric lineages of solenodons and hutias are interpreted as representing separate subspecies rather than distinct species, because they exhibit limited (albeit statistically detectable) morphometric differentiation [18, 63], in marked contrast to the substantial body size variance seen between Hispaniolan Nesophontes species that co-occur in the same landscapes. Similar patterns of allopatric differentiation and high island-wide beta diversity are also seen in several other Hispaniolan vertebrate taxa, including bats [64], birds [65] and reptiles [66]. Different components of Hispaniola’s mammal fauna therefore appear to have responded in different ways to Late Quaternary environmental change, exhibiting drastically different biogeographic patterns and rates of morphological change. Studying only the surviving representatives of this remarkable insular mammal fauna is thus unable to reveal the full scope of the evolutionary patterns and processes that generated regional Caribbean diversity.
We were able to compare ancient DNA data for the largest-bodied and smallest-bodied representatives of Hispaniola’s radiation of Nesophontes species, and the rapid rate of morphological evolution that we demonstrate in our palaeogenomic analyses of Hispaniolan Nesophontes is remarkable. It remains difficult to contrast this evolutionary rate against patterns seen more widely across the Eulipotyphla, because although this is one of the most species-rich mammalian orders, with 530 currently recognised extant species [24], relatively few comparative molecular studies investigating the phylogenetic relationships across other eulipotyphlan clades are available, and relatively few of these available studies have estimated divergence times between sister species rather than between higher-order groupings [67,68,69,70]. Greater interspecific variation in body mass compared to that between Hispaniolan Nesophontes species is seen within several morphologically and ecologically broadly comparable genera of soricids (“true shrews”), such as Sorex (smallest, S. minutissimus, 1.4–4 g; largest, S. bendirii, 10–21 g) and Suncus (smallest, S. malayanus, 1.1–2.4 g; largest, S. murinus, 23.5–147.3 g) [24]. However, small-bodied and large-bodied species are not closely related to each other within these soricid genera, in contrast to representing rapid recent evolutionary divergence of body sizes as seen in Hispaniolan Nesophontes [71, 72].
The rapid morphological change observed between N. paramicrus and N. zamicrus is instead more characteristic of evolutionary rates seen in other insular taxa. Island species adapt to novel conditions found in insular environments (e.g., reduced interspecific competition, resource limitation) through changes in size and morphology that are often rapid and large-scale, driven by processes such as competitive character displacement and character release [10, 13, 73, 74]. Evolutionary rates for insular mammals are shown to be accelerated in comparison with mainland species, with faster evolution not only a response to initial colonisation but also occurring over thousands of years [11]. However, the limited number of studies of body size change in extinct island vertebrates have focused on ‘charismatic’ large-bodied species [75,76,77], and our study therefore provides an important new example of rapid body size change in a now-extinct small-bodied insular vertebrate lineage.
Conclusions
Despite adverse preservational conditions for ancient biomolecules in Caribbean environments, we were able to recover aDNA successfully from the extinct Hispaniolan island-shrew Nesophontes zamicrus, the smallest known non-volant endemic Caribbean land mammal [14]. This is only the second extinct Caribbean eulipotyphlan species for which genomic data are available, and our data represent some of the first aDNA sequences that have been recovered for any extinct vertebrates known from the rich Holocene subfossil record of the Greater Antilles [78, 79].
Although it has been suggested that even the largest Caribbean islands only supported minimal species richness of endemic nesophontids [29], our combined morphometric and genetic analyses together demonstrate that Hispaniola supported a diverse late Quaternary nesophontid fauna comprising three co-occurring species, and that Nesophontes paramicrus and N. zamicrus diverged through rapid morphological evolution. The Late Quaternary Caribbean land mammal fauna thus consisted not only of ancient evolutionary lineages, such as the eulipotyphlan suborder Solenodonota and its two endemic families Nesophontidae and Solenodontidae [22, 80], but also recent evolutionary radiations that may have been driven by Quaternary environmental change, with Hispaniola representing a cradle of diversity as well as a museum of diversity [81]. This pattern of diversification is comparable to that seen on other ancient island terranes such as New Zealand, which also contains ancient vertebrate lineages such as kiwis (Apterygidae) that have similarly undergone recent species radiations [82, 83]. The insular Caribbean was therefore an important hotspot for the ongoing generation of new mammal species and evolutionary novelty, until the recent loss of much of this diversity following human arrival.
Methods
Morphometric analysis
We measured mandibles of adult Nesophontes specimens (defined as individuals showing complete dental eruption) in the Vertebrate Paleontology Collection, Florida Museum of Natural History (47 specimens) and the Palaeontology Collections, Natural History Museum, London (18 specimens) (Table S2). We took 13 measurements on each specimen (Table S1), using digital calipers accurate to 0.02 mm.
All statistical analyses were carried out in R v 6.3.0 [84]. We conducted PCA on our measurement dataset using the prcomp() function in R, first using raw linear data, and then using log-transformed measurements to account for the potential confounding influence of variation in size [85]. We used the ‘mclust’ package in R [86] to conduct hierarchical model-based cluster identification using principal component loadings, and determine if mandibles grouped into discrete clusters. We sequentially added principal component loadings until a distinct pattern of cluster classification was observed. Cluster models were unsupervised to ensure no bias in specimen classification, entailing no prior assignment of any specimens to a specific cluster, or constraint on the maximum number of potential clusters (to a maximum of nine by default in ‘mclust’). We then conducted ANOVAs to assess variation in each individual mandibular measurement between clusters, and conducted Tukey post-hoc tests to describe differences between clusters.
Ancient DNA analysis
We sampled seven Nesophontes partial crania from Cueva de Bosque Humido, identified as N. hypomicrus (n = 2) and N. zamicrus (n = 5) based on morphometric criteria given in ref. [28], by powdering the bone using a Mikro Dismembrator. We conducted extractions and Next Generation Sequencing library builds in a dedicated aDNA laboratory at the Natural History Museum, London. Each sample was processed using utensils cleaned with bleach and UV-treated before and after use, in order to limit cross-contamination. Extraction protocol followed ref. [87] and included the use of proteinase K for bone digestion and silica-spin columns for DNA purification. Single-index double-stranded DNA libraries were built following protocols in ref. [88]. Negative extraction and library-build controls were included during each process.
Nesophontes samples were screened using the NHM’s Illumina NextSeq 500 to assess DNA quality. Reads were de-multiplexed by index, adapters were removed, and paired end reads were quality checked and merged using default settings on CLC genomics workbench v.8 (CLC Bio-Qiagen, Aarhus, Denmark). Reads were then mapped using CLC genomics workbench to the previously-sequenced N. paramicrus whole-mitochondrial genome and nuclear genes (Table 1, Fig. S1). Mapping parameters were as follows: length fraction = 0.8, similarity fraction = 0.8. The highest-quality sample (based on number of reads mapping to the N. paramicrus mitochondrial genome; estimated endogenous content = 0.633%) was sent to the Department of Bioinformatics and Genetics at the Swedish Museum of Natural History for deeper sequencing on an Illumina HiSeq X. We concatenated the resulting whole-mitochondrial genome and nuclear genes [89, 90], and aligned our sample with data from GenBank for 25 extant eulipotyphlan genera and 10 eutherian mammal outgroup taxa (Table S3). We applied PartitionFinder [91] to choose the most appropriate partitioning scheme and best-fit evolutionary models (Table S4).
We constructed Bayesian trees using MrBayes [92] with four chains (three heated, one cold) that were run for 1 × 106 generations, sampling every 1 × 103 generations with a burn-in period of 250 trees. We generated a maximum likelihood tree with bootstrap support values using RAxML v.8 [93] implemented in CIPRES Science Gateway v.3 [94, 95]. We conducted divergence dating using fossil calibrations and priors taken from refs [96, 97] (Table S5), jointly estimating phylogeny and divergence dates under an uncorrelated relaxed lognormal clock [98]. Due to convergence issues in BEAST for large-scale genomics datasets, we employed the Hasegawa-Kishino-Yano (HKY) model of sequence evolution [99] with a gamma distribution of rates across sites. We used a Yule model of speciation; we also ran a birth-death model for comparison and generated an identical topology. Taxa sets for all monophyletic clades (including both outgroup and ingroup taxa) were constrained. We then applied lognormal priors based on available fossil data (Table S3) to seven taxa sets, with the remaining taxa sets left as the default priors. Clock rate priors were set to uninformative uniform distributions (upper = E100, lower = E12). We left all other priors as default values in BEAUti v.1.8.3 [100]. We ran analyses for 25 million generations, sampling every 1000 generations. We used Tracer v.1.6.0 (beast.community/) to assess convergence and effective sample size for all parameters after a burn-in of 10%. We generated a maximum credibility tree in TreeAnnotator v.1.8.3 [98], using trees sampled in the prior distribution.
Pairwise genetic distances between Nesophontes species were estimated using MEGA v.4 [101]. We used four nuclear genes (BDNF, BRCA1, CREM, RAG1) and two mitochondrial genes (cyt b, 12S) for genetic pairwise distance analyses (Table S3). We then determined pairwise sequence divergences (calculated as Kimura two-parameter distances) for Hispaniolan Nesophontes species, and also comparatively for congeneric pairs/groupings of 49 extant eulipotyphlan species currently recognised as valid [24], in 11 genera, using data from GenBank (Table S6).
Availability of data and materials
The DNA sequence data generated during the current study are available in the European Nucleotide Archive (accession number: PRJEB39675). Additional datasets supporting this paper are available in the Supplementary Information.
Abbreviations
- aDNA:
-
Ancient DNA
- ADORA3:
-
Adenosine A3 receptor
- ADRAB2:
-
Alpha 2B adrenergic receptor
- ADRA2B:
-
Alpha 2B adrenoceptor
- APOB:
-
Apolipoprotein B
- APP:
-
Amyloid beta precursor protein
- BDNF:
-
Brain-derived neurotrophic factor
- BIC:
-
Bayesian Information Criterion
- BMI1:
-
Polycomb complex protein BMI-1
- BRCA1:
-
Breast Cancer 1
- BP:
-
Years before present
- bp:
-
Base pairs
- CREM:
-
cAMP responsive element modulator
- cyt b :
-
Cytochrome b
- HPD:
-
Highest posterior density
- Ma:
-
Million years ago
- MY:
-
Million years
- PCA:
-
Principal component analysis
- PC1:
-
Principal component axis 1
- PLCB4:
-
Phospholipase C Beta 4
- RAG1:
-
Recombination activating gene 1
- 12S:
-
12S ribosomal RNA
References
Bunce M, Worthy TH, Ford T, Hoppitt W, Willerslev E, Drummond A, Cooper A. Extreme reversed sexual size dimorphism in the extinct New Zealand moa Dinornis. Nature. 2003;425:172–5.
Steeves TE, Holdaway RN, Hale ML, McLay E, McAllan IAW, Christian M, Hauber ME, Bunce M. Merging ancient and modern DNA: extinct seabird taxon rediscovered in the North Tasman Sea. Biol Lett. 2010;6:94–7.
Grosser S, Rawlence NJ, Anderson CNK, Smith IWG, Scofield RP, Waters JM. Invader or resident? Ancient-DNA reveals rapid species turnover in New Zealand little penguins. Proc R Soc B. 2016;283:20152879.
Turvey ST, Hansford J, Brace S, Mullin V, Gu S, Sun G. Holocene range collapse of giant muntjacs and pseudo-endemism in the Annamite large mammal fauna. J Biogeogr. 2016;43:2250–60.
Leffler EM, Bullaughey K, Matute DR, Meyer WK, Ségurel L, Venkat A, Andolfatto P, Przeworski M. Revisiting an old riddle: what determines genetic diversity levels within species? PLoS One. 2012;10(9):e1001388.
Romiguier J, et al. Comparative population genomics in animals uncovers the determinants of genetic diversity. Nature. 2014;515:261–3.
Darwin C. On the origin of species. London: John Murray; 1859.
Schluter D. The ecology of adaptive radiation. Oxford: Oxford University Press; 2000.
Losos JB, Ricklefs RE. Adaptation and diversification on islands. Nature. 2009;457:830–6.
Lomolino MV, van der Geer AA, Lyras GA, Palombo MR, Sax DF, Rozzi R. Of mice and mammoths: generality and antiquity of the island rule. J Biogeogr. 2013;40:1427–39.
Millien V. Morphological evolution is accelerated among island mammals. PLoS Biol. 2006;4:e321.
Turvey ST. Holocene extinctions. Oxford: Oxford University Press; 2009.
van der Geer A, Lyras G, de Vos J, Dermitzakis M. Evolution of island mammals: adaptation and extinction of placental mammals on islands. Oxford: Wiley-Blackwell; 2010.
Turvey ST, Fritz SA. The ghosts of mammals past: biological and geographical patterns of global mammalian extinction across the Holocene. Philos Trans R Soc Lond B. 2011;366:2564–76.
Ricklefs R, Bermingham E. The West Indies as a laboratory of biogeography and evolution. Philos Trans R Soc Lond B. 2008;363:2393–413.
Cooke SB, Dávalos LM, Mychajliw AM, Turvey ST, Upham NS. Anthropogenic extinction dominates Holocene declines of West Indian mammals. Annu Rev Ecol Evol Syst. 2017;48:301–27.
Díaz-Franco S. Situación taxonómica de Geocapromys megas (Rodentia: Capromyidae). Caribb J Sci. 2001;37:72–80.
Hansford J, Nuñez-Miño JM, Young RP, Brace S, Brocca JL, Turvey ST. Taxonomy-testing and the ‘Goldilocks Hypothesis’: morphometric analysis of species diversity in living and extinct Hispaniolan hutias. Syst Biodivers. 2012;10:491–507.
Turvey ST, Cooper JH. The past is another country: is evidence for prehistoric, historical and present-day extinction really comparable? In: Turvey ST, editor. Holocene extinctions. Oxford: Oxford University Press; 2009. p. 193–212.
Turvey ST, Weksler M, Morris EL, Nokkert M. Taxonomy, phylogeny and diversity of the extinct Lesser Antillean rice rats (Sigmodontinae: Oryzomyini), with description of a new genus and species. Zool J Linnean Soc. 2010;160:748–72.
Brace S, Turvey ST, Weksler M, Hoogland MLP, Barnes I. Unexpected evolutionary diversity in a recently extinct Caribbean mammal radiation. Proc R Soc B. 2015;282:20142371.
Brace S, Thomas JA, Dalén L, Burger J, MacPhee RDE, Barnes I, Turvey ST. Evolutionary history of the Nesophontidae, the last unplaced Recent mammal family. Mol Biol Evol. 2016;33:3095–103.
Kehlmaier C, Barlow A, Hastings AK, Vamberger M, Paijmans JLA, Steadman DW, Albury NA, Franz R, Hofreiter M, Fritz U. Tropical ancient DNA reveals relationships of the extinct Bahamian giant tortoise Chelonoidis alburyorum. Proc R Soc B. 2017;284:20162235.
Wilson DE, Mittermeier RA, editors. Handbook of the mammals of the world. Vol. 8. Insectivores, sloths and colugos. Barcelona: Lynx Edicions; 2018.
MacPhee RDE, Flemming C, Lunde DP. “Last occurrence” of the Antillean insectivoran Nesophontes: new radiometric dates and their interpretation. Am Mus Novit. 1999;3621:1–19.
Turvey ST, Oliver JR, Narganes Storde Y, Rye P. Late Holocene extinction of Puerto Rican native land mammals. Biol Lett. 2007;3:193–6.
Anthony HE. Preliminary diagnosis of an apparently new family of insectivores. Bull Am Mus Nat Hist. 1916;35:725–8.
Miller GS. A second collection of mammals from caves near St. Michel, Haiti. Smithson. Misc Collect. 1929;81(9):1–30.
Silva Taboada G, Suárez Duque W, Díaz FS. Compendio de los mamíferos terrestres autóctonos de Cuba vivientes y extinguidos. Museo Nacional de Historia Natural: La Habana; 2007.
Morgan GS, MacPhee RDE, Woods R, Turvey ST. Late Quaternary fossil mammals from the Cayman Islands, West Indies. Bull Am Mus Nat Hist. 2019;428:1–79.
Condis Fernández MM, Jiménez Vásquez O, Arredondo C. Revisión taxonómica del género Nesophontes (Insectivora: Nesophontidae) en Cuba: análisis de los caracteres diagnóstico. Monogr Soc Hist Nat Balears. 2005;12:95–100.
Choate JR, Birney EC. Sub-recent Insectivora and Chiroptera from Puerto Rico, with the description of a new bat of the genus Stenoderma. J Mammal. 1968;49:400–12.
McFarlane DA. A note on dimorphism in Nesophontes edithae (Mammalia: Insectivora), an extinct island-shrew from Puerto Rico. Caribb J Sci. 1999;35:142–3.
Bloch JI, Rose KD, Gingerich PD. New species of Batodonoides (Lipotyphla, Geolabididae) from the early Eocene of Wyoming: smallest known mammal? J Mammal. 1998;79:804–27.
Miller GS. Mammals eaten by Indians, owls, and Spaniards in the coast region of the Dominican Republic. Smithson. Misc Collect. 1929;82(5):1–16.
Miller GS. Three small collections of mammals from Hispaniola. Smithson. Misc Collect. 1930;82(15):1–10.
Woods CA, Ottenwalder JA. The natural history of southern Haiti. Gainesville: Florida Museum of Natural History; 1992.
Belando AL. El arte rupestre en el parque nacional los Haitises. 2009. http://www.rupestreweb.info/haitises2.html. Accessed 20 December 2019.
Bradley RD, Baker RJ. A test of the genetic species concept: cytochrome-b sequences and mammals. J Mammal. 2001;82:960–73.
González P, Sawyer YE, Avila M, Armién AG, Armién B, Cook JA. Variation in cytochrome-b haplotypes suggests a new species of Zygodontomys (Rodentia: Cricetidae) endemic to Isla Coiba, Panama. Zoologia. 2010;27:660–5.
Bannikova AA, Abramov AV, Borisenko AV, Lebedev VS, Rozhnov VV. Mitochondrial diversity of the white-toothed shrews (Mammalia, Eulipotyphla, Crocidura) in Vietnam. Zootaxa. 2011;2812:1–20.
Shinohara A, Kawada S, Son NT, Koshimoto C, Endo H, Can DN, Suzuki H. Molecular phylogeny of east and southeast Asian fossorial moles (Lipotyphla, Talpidae). J Mammal. 2014;95:455–66.
Frankham R. Relationship of genetic variation to population size in wildlife. Conserv Biol. 1996;10:1500–8.
Frankham R. Do island populations have less genetic variation than mainland populations? Heredity. 1997;78:311–27.
Eldridge MD, King JM, Loupis AK, Spencer PB, Taylor AC, Pope LC, Hall GP. Unprecedented low levels of genetic variation and inbreeding depression in an island population of the black-footed rock-wallaby. Conserv Biol. 1999;13:531–41.
Frankham R. Genetics and conservation biology. C R Biol. 2003;326:S22–9.
He K, Chen J, Gould GC, Yamaguchi N, Ai H, Wang Y, Zhang Y, Jiang X. An estimation of Erinaceidae phylogeny: a combined analysis approach. PLoS One. 2012;7:e39304.
Sato JJ, Ohdachi SD, Echenique-Diaz LM, Borroto-Páez R, Begué-Quiala G, Delgado-Labañino JL, Gámez-Díez J, Alvarez-Lemus J, Nguyen ST, Yamaguchi N, Kita M. Molecular phylogenetic analysis of nuclear genes suggests a Cenozoic over-water dispersal origin for the Cuban solenodon. Sci Rep. 2016;6:31173.
Bannikova AA, Zemlemerova ED, Colangelo P, Sözen M, Sevindik M, Kidov AA, Dzuev AA, Kryštufek B, Lebedev VS. An underground burst of diversity – a new look at the phylogeny and taxonomy of the genus Talpa Linnaeus, 1758 (Mammalia: Talpidae) as revealed by nuclear and mitochondrial genes. Zool J Linnean Soc. 2015;175:930–48.
He K, Li Y, Brandley MC, Lin L, Wang Y, Zhang Y, Jiang X. A multi-locus phylogeny of Nectogalini shrews and influences of the paleoclimate on speciation and evolution. Mol Phylogenet Evol. 2010;56:734–46.
Dubey S, Zaitsev M, Cosson JF, Abdukadier A, Vogel P. Pliocene and Pleistocene diversification and multiple refugia in a Eurasian shrew (Crocidura suaveolens group). Mol Phylogenet Evol. 2006;38:635–47.
Esteva M, Cervantes FA, Brant SV, Cook JA. Molecular phylogeny of long-tailed shrews (genus Sorex) from México and Guatemala. Zootaxa. 2010;2615:47–65.
Avise JC, Walker D, Johns GC. Speciation durations and Pleistocene effects on vertebrate phylogeography. Proc R Soc B. 1998;265:1707–12.
Liu S, Lorenzen ED, Fumagalli M, Li B, Harris K, Xiong Z, Zhou L, Sand Korneliussen T, Somel M, Babbitt C, Wray G, Li J, He W, Wang Z, Fu W, Xiang X, Morgan CC, Doherty A, O’Connell MJ, McInerney JO, Born EW, Dalén L, Dietz R, Orlando L, Sonne C, Zhang G, Nielsen R, Willerslev E, Wang J. Population genomics reveal recent speciation and rapid evolutionary adaptation in polar bears. Cell. 2014;157:785–94.
Rowe KC, Aplin KP, Baverstock PR, Moritz C. Recent and rapid speciation with limited morphological disparity in the genus Rattus. Syst Biol. 2011;60:188–203.
Lister AM. The impact of Quaternary ice ages on mammalian evolution. Philos Trans R Soc Lond B. 2004;359:221–41.
Barnosky AD. Effects of Quaternary climatic change on speciation in mammals. J Mamm Evol. 2005;12:247–64.
Maurrasse F, Pierre-Louis R, Rigaud JG. Cenozoic facies distribution in the southern peninsula of Haiti and the Barahona peninsula, Dominican Republic, and its relations concerning tectonic evolution of the La Selle-Baoruco block. Caribb Geol Collected Contrib. 1980;9:1–24.
Mann P, Draper G, Lewis JF. An overview of the geologic and tectonic development of Hispaniola. In: Mann P, Draper G, Lewis JF, editors. Geologic and tectonic development of the North America–Caribbean Plate boundary in Hispaniola, vol. 262. Boulder: Geological Society of America Special Publication; 1991. p. 1–51.
Pregill GK, Olson SL. Zoogeography of West Indian vertebrates in relation to Pleistocene climatic cycles. Annu Rev Ecol Syst. 1981;12:75–98.
Tziperman E, Gildor H. On the mid-Pleistocene transition to 100-kyr glacial cycles and the asymmetry between glaciation and deglaciation times. Paleoceanography. 2003;18:1–8.
Brace S, Barnes I, Powell A, Pearson R, Woolaver LG, Thomas MG, Turvey ST. Population history of the Hispaniolan hutia Plagiodontia aedium (Rodentia: Capromyidae): testing the model of ancient differentiation on a geotectonically complex Caribbean island. Mol Ecol. 2012;21:2239–53.
Turvey ST, et al. Independent evolutionary histories in allopatric populations of a threatened Caribbean land mammal. Divers Distrib. 2016;22:589–602.
Lim BK, Loureiro LO, Upham NS, Brocca JL. Phylogeography of Dominican Republic bats and implications for systematic relationships in the Neotropics. J Mammal. 2017;98:986–93.
Sly ND, Townsend AK, Rimmer CC, Townsend JM, Latta SC, Lovette IJ. Ancient islands and modern invasions: disparate phylogeographic histories among Hispaniola’s endemic birds. Mol Ecol. 2011;20:5012–24.
Gifford ME, Powell R, Larson A, Gutberlet RL. Population structure and history of a phenotypically variable teiid lizard (Ameiva chrysolaema) from Hispaniola: the influence of a geologically complex island. Mol Phylogenet Evol. 2004;32:735–48.
Quérouil S, Hutterer R, Barrière P, Colyn M, Kerbis Peterhans JC, Verheyen E. Phylogeny and evolution of African shrews (Mammalia: Soricidae) inferred from 16s rRNA sequences. Mol Phylogenet Evol. 2001;20:185–95.
Ohdachi SD, Isawa MA, Nesterenko VA, Abe H, Masuda R, Haberl W. Molecular phylogenetics of Crocidura shrews (Insectivora) in east and Central Asia. J Mammal. 2004;85:396–403.
Bannikova AA, Lavrenchenko LA, Kramerov DA. Phylogenetic relationships between Afrotropical and Palaearctic Crocidura species inferred from inter-SINE-PCR. Biochem Syst Ecol. 2005;33:45–59.
Dubey S, Salamin N, Ohdachi SD, Barrière P, Vogel P. Molecular phylogenetics of shrews (Mammalia: Soricidae) reveal timing of transcontinental colonizations. Mol Phylogenet Evol. 2007;44:126–37.
Omar H, Adamson EAS, Bhassu S, Goodman SM, Soarimalala V, Hashim R, Ruedi M. Phylogenetic relationships of Malayan and Malagasy pygmy shrews of the genus Suncus (Soricomorpha: Soricidae) inferred from mitochondrial cytochrome b gene sequences. Raffles Bull Zool. 2011;59:237–43.
Stanchak KE, Santana SE. Do ecogeographical rules explain morphological variation in a diverse, Holarctic genus of small mammals? J Biogeogr. 2019;46:110–22.
Dayan T, Simberloff D. Size patterns among competitors: ecological character displacement and character release in mammals, with special reference to island populations. Mammal Rev. 1998;28:99–124.
Yom-Tov Y, Yom-Tov S, Moller H. Competition, coexistence, and adaptation amongst rodent invaders to Pacific and New Zealand islands. J Biogeogr. 1999;26:947–58.
Paxinos EE, James HF, Olson SL, Sorenson MD, Jackson J, Fleischer RC. mtDNA from fossils reveals a radiation of Hawaiian geese recently derived from the Canada goose (Branta canadensis). Proc Natl Acad Sci USA. 2002;99:1399–404.
Bunce M, Szulkin M, Lerner HRL, Barnes I, Shapiro B, Cooper A, Holdaway RN. Ancient DNA provides new insights into the evolutionary history of New Zealand's extinct giant eagle. PLoS Biol. 2005;3:e9.
Meijer HJM, van den Hoek Ostende LW, van den Bergh GD, de Vos J. The fellowship of the hobbit: the fauna surrounding Homo floresiensis. J Biogeogr. 2010;37:995–1006.
Woods R, Turvey ST, Brace S, MacPhee RDE, Barnes I. Ancient DNA of the extinct Jamaican monkey Xenothrix reveals extreme insular change within a morphologically conservative primate radiation. Proc Natl Acad Sci U S A. 2018;115:12769–74.
Delsuc F, et al. Ancient mitogenomes reveal the evolutionary history and biogeography of sloths. Curr Biol. 2019;29:P2031–42.
Collen B, Turvey ST, Waterman C, Meredith HMR, Kuhn T, Baillie JEM, Isaac NJB. Investing in evolutionary history: implementing a phylogenetic approach for mammal conservation. Philos Trans R Soc Lond B. 2011;366:2611–22.
Mace GM, Gittleman JL, Purvis A. Preserving the tree of life. Science. 2003;300:1707–9.
Baker AJ, Daugherty CH, Colbourne R, McLennan JL. Flightless brown kiwis of New Zealand possess extremely subdivided population structure and cryptic species like small mammals. Proc Natl Acad Sci U S A. 1995;92:8254–8.
Shepherd LD, Lambert DM. Ancient DNA and conservation: lessons from the endangered kiwi of New Zealand. Mol Ecol. 2008;17:2174–84.
R Development Core Team. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2019.
Somers KM. Multivariate allometry and removal of size with principal components analysis. Syst Biol. 1986;35:359–68.
Scrucca L, Fop M, Murphy TB, Raftery AE. mclust 5: clustering, classification and density estimation using Gaussian finite mixture models. R J. 2016;8:205–33.
Dabney J, et al. Complete mitochondrial genome sequence of a middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc Natl Acad Sci U S A. 2013;110:15758–63.
Meyer M, Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc. 2010; pdb.prot5448.
Kearse M, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9.
Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8.
Lanfear R, Calcott B, Ho SY, Guindon S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 2012;29:1695–701.
Ronquist F, Huelsenbeck JP. MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–4.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML web servers. Syst Biol. 2008;57:758–71.
Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Proceedings of the Gateway Computing Environments Workshop (GCE), 2010. www.phylo.org/sub_sections/portal/sc2010_paper.pdf. Accessed 20 December 2019.
Benton MJ, Donoghue PCJ, Asher RJ. Calibrating and constraining molecular clocks. In: Hedges SB, Kumar S, editors. The timetree of life. Oxford: Oxford University Press; 2009. p. 35–86.
dos Reis M, Inoue J, Hasegawa M, Asher RJ, Donoghue PCJ, Yang Z. Phylogenomic datasets provide both precision and accuracy in estimating the timescale of placental mammal phylogeny. Proc R Soc B. 2012;279:3491–500.
Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214.
Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22:160–74.
Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST. Mol Biol Evol. 2012;29:1969–73.
Tamura K, Dudley J, Nei M, Kumar S. Molecular Revolutionary Genetic Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007;24:1597–9.
Acknowledgements
Field collection of subfossil material was assisted by Paul Scofield, Helen Meredith and Jorge Brocca, and access to museum collections was made possible by Richard Hulbert (Florida Museum of Natural History). The authors would like to acknowledge support from the Science for Life Laboratory, the National Genomics Infrastructure (NGI), Sweden, the Knut and Alice Wallenberg Foundation and UPPMAX for providing assistance in massively parallel DNA sequencing and computational infrastructure.
Funding
The study was financially supported by the Natural Environment Research Council (NE/L501803/1), the Royal Society (RG100902, UF130573), and the Biotechnology and Biological Sciences Research Council (BB/M009122/1). The funding bodies for this study had no role in study design, collection of data, data analysis and interpretation, or writing the manuscript.
Author information
Authors and Affiliations
Contributions
STT, IB, RW, EJR and CVM designed research. RW, CVM, IB, SB, STT and MFJB coordinated data collection. RW, SB, IB, CVM, LD, EJR and STT interpreted and analysed data. RW, STT, IB and CVM wrote the paper. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
(a) Distribution of read lengths in paired-reads dataset. (b) Level of coverage across reference mitochondrial genome in mapped-reads dataset. Figure S2. Maximum likelihood phylogeny generated using RAxML, including Nesophontes paramicrus, N. zamicrus, and extant eulipotyphlans with available whole mitochondrial genome data. Node values represent bootstrap support (100 replicates). Table S1. Morphometric measurements taken on Hispaniolan Nesophontes mandibles. Table S2. Measurement and PCA data for Hispaniolan Nesophontes mandibles. Table S3. Genes used in phylogenetic analysis, including GenBank accession numbers. Asterisk indicates chimeric taxon made up of multiple species. Table S4. Evolutionary models chosen for each gene in the alignment using PartitionFinder. Table S5. Fossil constraints and priors used in divergence date analysis. Table S6. Eulipotyphlan species used in pairwise genetic distance analysis. Table S7. Pairwise distances for eulipotyphlan sister species pairs in the mitochondrial cyt b gene, showing number of base differences per site between sequences. Table S8. Pairwise distances for eulipotyphlan sister species pairs in the mitochondrial 12S gene, showing number of base differences per site between sequences. Table S9. Pairwise distances for eulipotyphlan sister species pairs in the CREM (cAMP responsive element modulator) protein-coding nuclear gene, showing number of base differences per site between sequences. Table S10. Pairwise distances for eulipotyphlan sister species pairs in the BRCA1 (Breast Cancer 1) protein-coding nuclear gene, showing number of base differences per site between sequences. Table S11. Pairwise distances for eulipotyphlan sister species pairs in the RAG1 (recombination activating gene 1) protein-coding nuclear gene, showing number of base differences per site between sequences. Table S12. Pairwise distances for eulipotyphlan sister species pairs in the BDNF (brain derived neurotrophic factor) protein-coding nuclear gene, showing number of base differences per site between sequences.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Woods, R., Turvey, S.T., Brace, S. et al. Rapid size change associated with intra-island evolutionary radiation in extinct Caribbean “island-shrews”. BMC Evol Biol 20, 106 (2020). https://doi.org/10.1186/s12862-020-01668-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-020-01668-7