
Abstract
Background
The hypothesis that vertebrates have experienced two ancient, whole genome duplications (WGDs) is of central interest to evolutionary biology and has been implicated in evolution of developmental complexity. Three-way and Four-way paralogy regions in human and other vertebrate genomes are considered as vital evidence to support this hypothesis. Alternatively, it has been proposed that such paralogy regions are created by small-scale duplications that occurred at different intervals over the evolution of life.
Results
To address this debate, the present study investigates the evolutionary history of multigene families with at least three-fold representation on human chromosomes 1, 2, 8 and 20. Phylogenetic analysis and the tree topology comparisons classified the members of 36 multigene families into four distinct co-duplicated groups. Gene families falling within the same co-duplicated group might have duplicated together, whereas genes belong to different co-duplicated groups might have distinct evolutionary origins.
Conclusion
Taken together with previous investigations, the current study yielded no proof in favor of WGDs hypothesis. Rather, it appears that the vertebrate genome evolved as a result of small-scale duplication events, that cover the entire span of the animals’ history.
Similar content being viewed by others

Background
To elucidate the genetic underpinnings of major changes in organismal make up and the origination of ample new traits during the evolutionary history of vertebrates, Susumu Ohno in the year 1970 put forward the hypothesis that two rounds of whole genome duplications (WGDs) occurred early in vertebrate evolution. This hypothesis is popularly termed as “2R hypothesis” (two rounds of WGDs) and is believed to be the most rational explanation for the complexity of modern-day vertebrate genome [1]. The 2R has been under immense scrutiny over the past couple of decades [2,3,4,5,6,7,8,9]. The occurrence of intra-genomic conserved syntenic blocks (paralogy groups/paralogons) in vertebrate genomes is presented as the most credible proof furthering the ancient WGDs [10, 11]. Markedly, the presence of four potential quadruplicated regions on Homo sapiens autosomes (Hsa) 1/6/9/19 (MHC bearing paralogon), Hsa 4/5/8/10 (FGFR bearing chromosomes), Hsa 1/2/8/20 and Hsa 2/7/12/17 (HOX-cluster bearing chromosomes), is considered as an outcome of two consecutive rounds of WGDs [12]. However, alternatively it is hypothesized that the excess of paralogy regions in the human and other vertebrate genomes is due to higher instance of local duplications, translocations and chromosomal restructuring that occurred extensively at different intervals during early vertebrate history, thus nullifying the Ohno’s postulation [13].
In order to evaluate the mechanisms behind the formation of vertebrate paralogy regions, our research group has continuously been putting efforts in assembling and dating the gene duplications that occurred during the animal’s evolutionary history [3, 4, 7, 14,15,16,17]. Previously, we investigated the evolutionary histories of 11 multigene families (40 human genes) with triplicated or quadruplicated presence on Hsa 1/2/8/20. The results achieved were in contrast with 2R hypothesis, suggesting that the paralogy fragments on human chromosomes 1, 2, 8 and 20 are an outcome of small-scale duplication events which scattered across the history of metazoans [3, 4, 14, 17, 18].
In this study, we furthered our efforts [14] to analyze the evolutionary history of 25 human multigene families with three or fourfold distribution on Hsa 1/2/8/20. A robust and detailed phylogenomic analysis was carried out by using the recently available well-annotated and high-quality genome sequence data from a wide range of metazoans [19,20,21]. The topology comparison approach was particularly applied on the phylogenetic data of total 36 families (25 present data and 11 previous data) to classify the genes that might have duplicated together early in vertebrate history [3, 14]. In addition, relative timing approach was employed to estimate the timings of gene duplication events. In sync with the previous results [14], it appeared that the triplicated or quadruplicated gene families residing on Hsa 1/2/8/20 have not arisen simultaneously through 2R. Rather, phylogenetic data clarifies that the tetra-paralogy blocks on the human genome have resulted from independent duplications, segmental duplications and genomic restructuring events that had occurred at broadly different time points during the course of animal evolution.
Results
For investigating the validity of whole genome duplications (WGDs) hypothesis, which strongly supports that fourfold paralogons in the human genome had been formed by polyploidization events, we undertook phylogenetic analyses for 25 gene families (see details in Methods). Each of these chosen subset of multigene families have at least threefold portrayal on one of the paralogy regions in human genome that comprises of segments from human chromosomes 1, 2, 8 and 20 (Fig. 1; Table 1). By employing currently available wide range of sequenced vertebrate and invertebrate genomes, orthologous sequence data was gathered. (Additional file 1). This wider set of taxonomic representation in the sequence data enabled us to perform a robust phylogenetic examination based on NJ and ML methods (Additional files 2, 3 and 4). Given the phylogenetic data, we next determine the co-duplication events by employing the topology comparison approach [3, 17, 22] (Fig. 2). The phylogenetic tree topology comparison approach takes into account uniformity among tree branching pattern of distinct but physically linked gene families as a proof of their joint origin, thus displaying co-duplicated groups [13, 23]. In contrast, the non-uniform tree topologies of physically linked distinct families suggest the incongruent duplication histories of concerned genes [16]. For this purpose, only those sections of 36 phylogenies were chosen for which there is a strong bootstrap support for at least two gene duplication events within the time frame that divided the teleosts and vertebrates from tetrapods and invertebrates respectively (proposed timing of WGDs) (Additional file 5: Table S1). Among them 11 families were published previously by our research group [14].

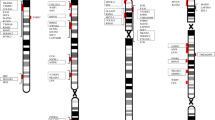
Evolutionary history of human tetra-paralogon Hsa 1/2/8/20. A circular view of human chromosomes shows the paralogons detected among human chromosomes 1/2/8/20, including the synteny relationship among 36 distinct multigene families: 11 families from previously published data that are labeled in black [14], whereas the 25 families analyzed in the present study that are labeled in green. Blue lines connect positions on ideograms for gene families with 3-fold representation, while yellow lines connect families with four-fold representation on these chromosomes. Detailed information about each family is given in Table 1

The human genes duplicated in parallel lie in respective co-duplicated groups. Consistencies in phylogenetic tree topologies of families (analyzed in this and our previous study) with at least threefold representation on human tetra-paralogon Hsa1/2/8/20 (a) Schematic topology of MROH and STK families; b schematic topology of E2F, EYA and STMN families; c schematic topology of HCK, DLGAP, NKAIN, KCNQ and MATN gene families; d schematic topology of FAM110, NCO, KCNS, YTHDF, XKR and MYT gene families. For each case, the percentage bootstrap values of internal branches are provided in parentheses except for gene families exhibiting slightly lower bootstrap values (≤50%).The connecting bars on the left portray the close physical associations of relevant genes. Asterisk symbol * designate the relevant chromosomes
MROH and STK gene family members has threefold representation on Hsa 1/2/8/20 paralogon and diversified by at least two vertebrate specific duplication events (Additional file 2). Assuming three independent gene translocation events in STK gene family, congruent but asymmetrical topologies of the type ((Hsa20/2 Hsa1/13) Hsa8/X) are recovered for these two gene families (Fig. 2a). This pattern indicates that the subset members of MROH and STK families might have duplicated in block through segmental duplication (SD) events.
E2F family has fourfold representation, whereas EYA and STMN families has threefold representation on tetra-paralogon Hsa 1/2/8/20. Assuming two independent gene translocation events revealed congruent and asymmetrical topologies of the type (((Hsa1/6 Hsa8/6) Hsa20) for E2F, EYA and STMN families (Fig. 2b; Additional file 2).
MATN family has fourfold presense, whereas HCK, DLGAP, NKAIN and KCNQ families has threefold portrayal on tetra-paralogy regions residing on Hsa 1/2/8/20. By assuming five gene translocation events, congruent and symmetrical topology of the type ((A, B) (C, D)) i.e. ((Hsa20-Hsa8/18) (Hsa1-Hsa8/6/2)) is recovered for HCK, DLGAP, NKAIN, KCNQ, and MATN families (Fig. 2c; Additional file 2).
FAM110 family has fourfold depiction whereas NCOA, KCNS, YTHDF, XKR, and MYT families has threefold distribution on Hsa 1/2/8/20. Each of these five families experienced at least two vertebrate specific duplication events (Additional file 2). By assuming four independent gene translocation events, members of these five families constitute the fourth co-duplicated group with an asymmetrical tree topology of the type ((Hsa20-Hsa8/2) Hsa2/1/8) (Fig. 2d).
Phylogenetic trees of eight gene families (CHRN, RGS, GRHL, RIMS, RSPO, ID, TCEA, and SNT) involve complex histories with majority of duplications occurred anciently prior to vertebrate–invertebrate split. CHRN family appear to have diversified by in total twelve duplications, six of them predate the vertebrate-invertebrate split (Additional file 2). RGS family tree indicates 10 duplication events, five of them occurred earlier than vertebrate-invertebrate split (Additional file 2). The tree topology pattern of GRHL indicates in total six duplications, two of them occured at least prior to protostome–deuterostome split (Additional file 2). The tree topology of RIMS family reveals three duplication events, one of them occurred earlier than Bilaterian–Nonbilaterian divergence (Additional file 2). RSPO arose by three independent gene duplication events, one of them happened prior to the divergence of echinoderms from vertebrates (Additional file 2). Vertebrate ID family tree revealed three independent gene duplication events, two of them occurred prior to hemichordates-vertebrates split (Additional file 2). Members of TCEA family arose by four duplications, three of them occurred earlier than vertebrate-cephalochordate split (Additional file 2). SNT paralogs experienced five duplications, four of them occurred prior to protostomes and deuterostomes split (Additional file 2).
Phylogenetic tree topologies of five families (AZIN, CRO, SLC, SNX and UBXN) reveal no evidence for vertebrate specifc gene duplications. All of these families are diversified by duplications that predates the vertebrate-invertebrate split (Additional file 2).
Estimation of gene duplication events with respect to relative timing of speciations provides a bird’s eye view to all the duplications that occurred in a particular time window [24]. Taken together the phylogenetic histories of 36 families (25 present data and 11 previously analyzed); in total 172 duplication events are recovered (Fig. 3). It appears that 52 of these duplication events occurred earlier than invertebrate-vertebrate- split, whereas 74 duplications are identified at the root of vertebrate history prior to tetrapod-teleost- divergence. Furthermore, 42 teleost fish specific and only 4 tetrapod specific duplication events are detected (Fig. 3).

The relative timings of gene duplication events. For the 36 multigene families analyzed in this study, 52 gene duplications are detected before the invertebrate-vertebrate divide and 74 duplications are detected after invertebrate-vertebrate and before tetrapod-bony fish divergence. Only four tetrapod specific duplication events are detected. The numbers enclosed in the parentheses following gene family names represent the count of duplications experienced by family. Gene families are ordered alphabetically
Discussion
Different post genomic methods like, genome wide pairwise comparisons and genome self comparisons have been robustly utilized in order to analyze the evolutionary basis for the origination of paralogy blocks in vertebrate genomes [11]. Evolutionary events in the recent vertebrate history has been successfully highlighted by these approaches, as the identity of recently duplicated intra-genomic and inter-genomic conserved syntenic segments and thus the patterns of evolution preceeding their origin are not vagued by evolutionary divergence, and genomic anomalies like chromosomal breakage and rearrangements [25]. For instance, complex pattern of segmental duplications (SDs) has been witnessed as a result of inter-genomic and intra-genomic comparisons in primates [26,27,28,29]. These large duplicated segments range in size from 300 kb to 1 Mb, position on at least two different genomic locations and possess more than 90% sequence identity [30]. Comparative data has implicated numerous roles to these SDs, such as creating new genes, expanding gene families and catalyzing large-scale hominoid specific chromosomal reorganization [31].
Conflictingly, carrying out inter-genomic and intra-genomic map comparisons have not proven useful in prediction of evolutionary processes that have arisen in early vertebrate history [32]. The reason lies in the fact that anciently duplicated genomic blocks have undergone events such as sequence variation, multiple chromosomal breakages, gene rearrangement events and modification of karyotype [32].
Phylogenetic investigation of multigene families is considered as the most reliable approach to estimate the existence of ancient intra-genomic synteny blocks or paralogons [16]. Evolutionary mechanisms behind the origin of anciently duplicated regions are captured more adequately by this approach: firstly, by estimating the relative timing of gene duplication events. This startegy can provide a bird’s eye view to all the duplications that happened in a specific time frame. For example, if the phylogenies designate that the bulk of the paralogy regions arose before the split of teleost-tetrapod and after the vertebrate-invertebrate- divergence, this advocates that large-scale gene duplications have occurred between these speciation events [24]. Secondly, the creation of paralogy regions can be scrutinized by combining the information from the global physical structuring of gene families comprising of paralogons with their phylogenetic tree topologies [13]. Distinct but physically linked multigene families (bearing human paralogons) showing coherence among the topologies would suggest that these families might have arisen jointly through segmental duplication events. This approach is elaborated and applied in previous studies [7, 16, 23].
In the earlier studies, various human tetra-paralogons, e.g. Hsa 4/5/8/10 (FGFR-paralogon), Hsa 2/7/12/17 (HOX-paralogon), and Hsa 1/6/9/19 (MHC-paralogon) have been examined to test the legitimacy of 2R hypothesis [4, 7, 14, 17, 23]. In this study, we assess the history of one of the most extensively cited paralogy region, which involves segments of human chromosomes 1, 2, 8 and 20 [14] (Additional files 2, 3 and 4). Taken together with our previous findings, this study estimated the history of 36 multigene families (25 present study and 11 from previous work) with at least threefold distribution on Hsa 1/2/8/20 [14] (Fig. 1; Table 1). In total, our data for this particular human paralogon involves 165 human genes and 2240 protein sequences (Additional file 1) [14]. The topology comparison approach is applied to test the WGD hypothesis (Fig. 2). Hence, the careful analysis resulted in the categorization of 36 phylogenies into four distinct co-duplicated groups, where the component gene families were expanded through duplications that could have happened within the time frame of invertebrate-vertebrate and bony fish-tetrapod- divergence (Additional file 5: Table S1). Distinct gene families within a co-duplicated group could have diversified concurrently by segmental duplications, whereas distinct co-duplicated groups might have been created through discrete duplication events [13]. The retrieval of large co-duplicated groups in this study shows that ancient segmental duplications (aSDs) and rearrangement events played an essential role in modeling the paralogy segments belonging to human chromosomes 1/2/8/20 (Fig. 2). Interestingly, compatible and symmetrical topologies of the type ((AB) (CD)) are gained for the HCK, DLGAP, NKAIN, KCNQ, and MATN gene families (co-duplicated group 3) (Fig. 2c). This pattern is usually measured as an outcome of WGD events [12]. However, here we affirm that sub-chromosomal duplications might be a more balanced clarification for such symmetrical topology trends [6, 7, 14]. For example, tandem duplications occurring in two rounds embracing several unrelated genes would result in a genomic segment with specific paralogous gene-quartets organized in a tandem pattern. Genomic breakage of such larger segments into smaller subsegments via chromosomal deterioration and restructuring could result in paralogy blocks seen in human and other vertebrate genomes [14].
Conclusion
The present study examined the vertebrate polyploidy proposal by scrutinizing the phylogenomic history of human tetra-paralogon Hsa1/2/8/20. Estimation of gene duplication number with respect to speciation and topology comparison approach revealed no evidence in favor of Ohno’s 2R model. Instead, taken together with previous results from HOX paralogon [16] (63 gene families), FGFR paralogon [4] (80 gene families) and MHC paralogon [23] (40 gene families), the present data (36 families from Hsa 1/2/8/20) suggests that vertebrate genome in its early history was shaped by small-scale events, such as duplication of independent genes, chromosomal segments and rearrangements.
Methods
Data collection
Gene families with triplicated or quadruplicated presence on Hsa 1/2/8/20 were recognized by scanning the maps of human genome sequence at Ensembl genome browser [33,34,35]. A total of 25 gene-families (in total 125 known protein-coding genes) were identified. Among these gene families, 3 families have quadruplicated representation while the 22 families have triplicated presence on Hsa 1/2/8/20 (Fig. 1; Table 1).
The closest putative orthologs of human protein sequences in other animal species were acquired using BLASTP [36] in the Ensembl genome browser [33]. In attempts to obtain sequence data from those organisms still not available at Ensembl, a BLASTP search was carried out against the protein databases available at the National Center for Biotechnology Information [37] and the Joint Genome Institute [http://www.jgi.doe.gov/]. In total, 1605 amino acid sequences from 46 metazoan species were selected for phylogenomic investigation (Additional file 1). Further confirmation of the common ancestry of the putative orthologs was obtained by clustering homologous proteins within phylogenetic trees. The phylogenetic tree topology of each gene family was validated with the detailed comparison against a well established metazoan specie tree [38, 39]. Protein sequences whose placement within a tree was in disagreement with the conventional animal history were removed from the analysis.
The list of sequences used in the analysis (from 46 species including 25 tetrapods, 5 teleost fish, and 16 invertebrates) is provided in Additional file 1. The species that were selected for analysis included Homo sapiens (Human), Mus musculus (Mouse), Pan troglodytes (Chimpanzee), Gorilla gorilla (Gorilla), Callithrix jacchus (Marmoset), Pongo abelii (Orangutan), Macaca mulatta (Macaque), Rattus norvegicus (Rat), Oryctolagus cuniculus (Rabbit), Taeniopygia guttata (Zebra finch), Gallus gallus (Chicken), Canis familiaris (Dog), Felis catus (Cat), Bos taurus (Cow), Loxodonta Africana (Elephant), Equus caballus (Horse), Myotis lucifugus (Microbat), Dasypus novemcinctus (Armadillo), Pteropus vampyrus (Megabat), Ornithorhynchus anatinus (Platypus), Monodelphis domestica (Opossum), Pelodiscus sinensis (Chinese softshell turtle), Anolis carolinensis (Lizard), Erinaceus europaeus (Hedgehog), Xenopus tropicalis (Frog), Danio rerio (Zebrafish), Takifug urubripes (Fugu), Tetraodon nigroviridis (Tetraodon), Gasterosteus aculeatus (Stickleback), Oryzias latipes (Medaka), Branchiostoma floridae (Amphioxus), Ciona intestinalis (Ascidian), Ciona savignyi (Ascidian), Saccoglossus kowalevskii, Ptychodera flava, Strongylocentrotu spurpuratus (Sea urchin), Caenorhabditis elegans (Nematode), Anopheles gambiae (Mosquito), Drosophila melanogaster (Fruit fly), Apis mellifera (Honey bee), Capitella teleta (Capitella), Octopus bimaculoides (Octopus), Hydra magnipapillata (Hydra) and Nematostella vectensis (Sea anemone), Trichoplax adhaerens (Trichoplax), and Amphimedon queenslandica (Sponge).
Alignment and phylogenetic analysis
Phylogenetic analysis for each gene family was performed using MEGA version 5 [40]. Multiple sequence alignment program CLUSTALW [41] was used to align the protein sequences. Alignment quality has much impact on accurate inference of phylogeny. Homologous protein sequences often evolve under different evolutionary pressure in some regions of protein in different species [42,43,44]. Furthermore, regional rate heterogeneity affect the whole alignment and ultimaley phylogenetic reconstructuction [44, 45]. Therefore, multiple sequence alignment of each gene family was trimmed to eliminate all of positions containing gaps and missing data. Only unambiguous portions of sequence alignments are used for phylogenetic analyses. Phylogenetic analyses were performed using Neighbor-Joining (NJ) approach [46,47,48]. The JTT (Jones-Taylor-Thornton) matrix-based method and uncorrected proportion (p) of amino acid differences were employed as amino acid substitution models. Results obtained with both the methods are given in Additional files 2 and 3.The authenticity of clustering patterns in resulting trees was evaluated by bootstrap method (1000 pseudo-replicates) [49], which produced the bootstrap probability values for each interior branch in the phylogenetic tree. Each of the phylogenetic tree reconstruction methods has its own limitation, therefore, to systematically check and validate NJ based trees, Maximum Likelihood (ML) based phylogenies are also constructed using Whelan and Goldman (WAG) model of amino acid replacement [50]. The phylogenetic trees with the highest log likelihood scores are selected as final trees. Initial tree(s) for ML were generated automatically by applying NJ and BioNJ methods to a matrix of pairwise distances calculated using JTT model, and then selecting a toplogy with superior loglikelihood value [47, 51]. Heuristic searches starting with the initial trees were conducted with Nearest Neighbor Interchange [NNI] [40]. The topological reliability of each ML tree was evaluated by bootstrap method on the basis of 1000 pseudoreplicates [49]. The ML based trees are provided in Additional file 4.
The gene duplications relative to the divergence of major animal taxa were estimated by investigating the branching order of phylogenetic trees [4, 13, 18]. The phylogenetic topology of each family was compared with that of all other families to assess the consistencies in gene duplication events [16]. Gene families with consistent tree topologies are placed in respective co-duplicated groups [13].
Among the tree topologies of 25 gene families, the phylogenies of five families (MYT, NCOA, STMN, NKAIN and YTHDF) were rooted with invertebrate sequences, whereas CRO, ID, MROH, RSPO, FAM110, TCEA, RIMS, KCNQ and CHRN families were rooted with both invertebrate and vertebrate sequences. In case of UBXN and E2F families the vertebrate sequences served as outgroup. The phylogenies of SNX, RGS, GRHL, AZIN, DLGAP, STK, SLC, SNT, and XKR families contained two sub families, each of them served to root the other.
Availability of data and materials
The datasets analyzed during the current study are available in the Ensembl database (http://www.ensembl.org), NCBI database (https://www.ncbi.nlm.nih.gov/) and as supplementary information.
Abbreviations
- aSDs:
-
Ancient segmental duplications
- Hsa:
-
Homo sapiens autosomes
- JTT:
-
Jones-Taylor-Thornton
- ML:
-
Maximum Likelihood
- NJ:
-
Neighbor-Joining
- SDs:
-
Segmental duplications
- WAG:
-
Whelan and Goldman
- WGDs:
-
Whole genome duplications
References
Ohno S. Duplication for the sake of producing more of the same. In: Evolution by Gene Duplication. 1970: Springer Berlin Heidelberg. p. 59–65.
Abbasi AA. Are we degenerate tetraploids? More genomes, new facts. Biol Direct. 2008;3(1):1.
Abbasi AA. Unraveling ancient segmental duplication events in human genome by phylogenetic analysis of multigene families residing on HOX-cluster paralogons. Mol Phylogenet Evol. 2010;57(2):836–48.
Hafeez M, Shabbir M, Altaf F, Abbasi AA. Phylogenomic analysis reveals ancient segmental duplications in the human genome. Mol Phylogenet Evol. 2016;94:95–100.
Hughes AL, Friedman R. 2R or not 2R: testing hypotheses of genome duplication in early vertebrates. J Struct Funct Genom. 2003;3(1–4):85–93.
Abbasi AA. Piecemeal or big bangs: correlating the vertebrate evolution with proposed models of gene expansion events. Nat Rev Genet. 2010;11(2):166.
Ajmal W, Khan H, Abbasi AA. Phylogenetic investigation of human FGFR-bearing paralogons favors piecemeal duplication theory of vertebrate genome evolution. Mol Phylogenet Evol. 2014;81:49–60.
Sidow A. Gen (om) e duplications in the evolution of early vertebrates. Curr Opin Genet Dev. 1996;6(6):715–22.
Furlong RF, Holland PW. Were vertebrates octoploid? Philos Trans R Soc Lond B: Biol Sci. 2002;357(1420):531–44.
Gibson T, Spring J. Evidence in favour of ancient octaploidy in the vertebrate genome. Biochem Soc Trans. 2000;28(2):259–64.
McLysaght A, Hokamp K, Wolfe KH. Extensive genomic duplication during early chordate evolution. Nat Genet. 2002;31(2):200–4.
Lundin LG, Larhammar D, Hallbook F. Numerous groups of chromosomal regional paralogies strongly indicate two genome doublings at the root of the vertebrates. J Struct Funct Genomics 2003;3:53–63.
Hughes AL, da Silva J, Friedman R. Ancient genome duplications did not structure the human Hox-bearing chromosomes. Genome Res. 2001;11(5):771–80.
Abbasi AA, Hanif H. Phylogenetic history of paralogous gene quartets on human chromosomes 1, 2, 8 and 20 provides no evidence in favor of the vertebrate octoploidy hypothesis. Mol Phylogenet Evol. 2012;63(3):922–7.
Asrar Z, Haq F, Abbasi AA. Fourfold paralogy regions on human HOX-bearing chromosomes: role of ancient segmental duplications in the evolution of vertebrate genome. Mol Phylogenet Evol. 2013;66(3):737–47.
Ambreen S, Khalil F, Abbasi AA. Integrating large-scale phylogenetic datasets to dissect the ancient evolutionary history of vertebrate genome. Mol Phylogenet Evol. 2014;78:1–13.
Abbasi AA, Grzeschik K-H. An insight into the phylogenetic history of HOX linked gene families in vertebrates. BMC Evol Biol. 2007;7(1):1.
Martin A. Is tetralogy true? Lack of support for the “one-to-four rule”. Mol Biol Evol. 2001;18(1):89–93.
Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, Down T, et al. The Ensembl genome database project. Nucleic Acids Res. 2002;30(1):38–41.
Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–5.
Members BDC. Database resources of the BIG data center in 2019. Nucleic Acids Res. 2019;47(Database issue):D8.
Zhang J, Nei M. Evolution of Antennapedia-class homeobox genes. Genetics. 1996;142(1):295–303.
Naz R, Tahir S, Abbasi AA. An insight into the evolutionary history of human MHC paralogon. Mol Phylogenet Evol. 2017;110:1–6.
Van de Peer Y. Computational approaches to unveiling ancient genome duplications. Nat Rev Genet. 2004;5(10):752–63.
Dennis MY, Harshman L, Nelson BJ, Penn O, Cantsilieris S, Huddleston J, Antonacci F, Penewit K, Denman L, Raja A, et al. The evolution and population diversity of human-specific segmental duplications. Nat Ecol Evol. 2017;1(3):69.
Bailey JA, Eichler EE. Primate segmental duplications: crucibles of evolution, diversity and disease. Nat Rev Genet. 2006;7(7):552.
Cheng Z, Ventura M, She X, Khaitovich P, Graves T, Osoegawa K, Church D, DeJong P, Wilson RK, Paabo S, et al. A genome-wide comparison of recent chimpanzee and human segmental duplications. Nature. 2005;437(7055):88–93.
Feng X, Jiang J, Padhi A, Ning C, Fu J, Wang A, Mrode R, Liu JF. Characterization of genome-wide segmental duplications reveals a common genomic feature of association with immunity among domestic animals. BMC Genomics. 2017;18(1):293.
Zhao Q, Ma D, Vasseur L, You M. Segmental duplications: evolution and impact among the current Lepidoptera genomes. BMC Evol Biol. 2017;17(1):161.
Samonte RV, Eichler EE. Segmental duplications and the evolution of the primate genome. Nat Rev Genet. 2001;3:65–72.
Marques-Bonet T, Girirajan S, Eichler EE. The origins and impact of primate segmental duplications. Trends Genet. 2009;25(10):443–54.
Abbasi AA. Diversification of four human HOX gene clusters by step-wise evolution rather than ancient whole-genome duplications. Dev Genes Evol. 2015;225(6):353–7.
Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S. Ensembl 2015. Nucleic Acids Res. 2015;43(D1):D662–9.
Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, Billis K, Cummins C, Gall A, Girón CG, et al. Ensembl 2018. Nucleic Acids Res. 2018;46(D1):D754–61. https://doi.org/10.1093/nar/gkx1098.
Aken BL, Achuthan P, Akanni W, Amode MR, Bernsdorff F, Bhai J, Billis K, Carvalho-Silva D, Cummins C, Clapham P, et al. Ensembl 2017. Nucleic Acids Res. 2017;45(D1):D635–42.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10.
Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36(suppl 2):W5–9.
Adoutte A, Balavoine G, Lartillot N, Lespinet O, Prud'homme B, De Rosa R. The new animal phylogeny: reliability and implications. Proc Natl Acad Sci. 2000;97(9):4453–6.
Kumar S, Hedges SB. A molecular timescale for vertebrate evolution. Nature. 1998;392(6679):917.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–9.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–80.
Henikoff S, Henikoff JG. Protein family classification based on searching a database of blocks. Genomics. 1994;19(1):97–107.
Pesole G, Attimonelli M, Preparata G, Saccone C. A statistical method for detecting regions with different evolutionary dynamics in multialigned sequences. Mol Phylogenet Evol. 1992;1(2):91–6.
Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56(4):564–77.
Yang Z. On the best evolutionary rate for phylogenetic analysis. Syst Biol. 1998;47(1):125–33.
Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406–25.
Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Bioinformatics. 1992;8(3):275–82.
Russo C, Takezaki N, Nei M. Efficiencies of different genes and different tree-building methods in recovering a known vertebrate phylogeny. Mol Biol Evol. 1996;13(3):525–36.
Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–91.
Whelan S, Goldman N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol. 2001;18(5):691–9.
Gascuel O. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol Biol Evol. 1997;14(7):685–95.
Acknowledgements
The authors thank Yasir Mahmood Abbasi (computer programmer) for technical support.
Funding
This work was supported by National Key Research and Development Program of China [2016YFE0206600 to Y.B.]; International Partnership Program of the Chinese Academy of Sciences [153F11KYSB20160008 to Y.X.]; The 13th Five-year Informatization Plan of Chinese Academy of Sciences [XXH13505-05 to Y.B.]; The 100-Talent Program of Chinese Academy of Sciences [to Y.B. and Z.Z.]; The Open Biodiversity and Health Big Data Initiative of IUBS [to Y.B.]. The funding bodies had no role in the design of the study, collection, analysis, interpretation of data nor the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
AAA conceived the project. AAA and Y B designed the experiments. AQ and NS performed the experiments. AAA, YB, ZZ, YX, NS, AQ, RZ, SA, NP and NR analyzed the data. AAA, YB, NS, ZZ and AQ wrote the paper. It is also to affirm that all authors of the paper have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Complete list of protein sequences used in this study (PDF 1724 kb)
Additional file 2:
Neighbor Joining Trees of gene families (residing on human chromosomes 1/2/8/20) using p-distance method. (PDF 4993 kb)
Additional file 3:
Neighbor Joining Trees of gene families (residing on human chromosomes 1/2/8/20) using JTT method. (PDF 3402 kb)
Additional file 4:
Maximum likelihood Trees of gene families (residing on human chromosomes 1/2/8/20) based on WAG model. (PDF 3836 kb)
Additional file 5:
Table S1. Summary of the Phylogenetic analysis of gene families with three or more members are residing on human chromosomes 1/2/8/20. (PDF 73 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Pervaiz, N., Shakeel, N., Qasim, A. et al. Evolutionary history of the human multigene families reveals widespread gene duplications throughout the history of animals. BMC Evol Biol 19, 128 (2019). https://doi.org/10.1186/s12862-019-1441-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-019-1441-0