
Abstract
Background
Maternally inherited Wolbachia symbionts infect D. melanogaster populations worldwide. Infection rates vary greatly. Genetic diversity of Wolbachia in D. melanogaster can be subdivided into several closely related genotypes coinherited with certain mtDNA lineages. mtDNA haplotypes have the following global distribution pattern: mtDNA clade I is mostly found in North America, II and IV in Africa, III in Europe and Africa, V in Eurasia, VI is global but very rare, and VIII is found in Asia. The wMel Wolbachia genotype is predominant in D. melanogaster populations. However, according to the hypothesis of global Wolbachia replacement, the wMelCS genotype was predominant before the XX century when it was replaced by the wMel genotype. Here we analyse over 1500 fly isolates from the Palearctic region to evaluate the prevalence, genetic diversity and distribution pattrern of the Wolbachia symbiont, occurrence of mtDNA variants, and finally to discuss the Wolbachia genotype global replacement hypothesis.
Results
All studied Palearctic populations of D. melanogaster were infected with Wolbachia at a rate of 33–100%. We did not observe any significant correlation between infection rate and longitude or latitude. Five previously reported Wolbachia genotypes were found in Palearctic populations with a predominance of the wMel variant. The mtDNA haplotypes of the I_II_III clade and V clade were prevalent in Palearctic populations. To test the recent Wolbachia genotype replacement hypothesis, we examined three genomic regions of CS-like genotypes. Low genetic diversity was observed, only two haplotypes of the CS genotypes with a ‘CCG’ variant predominance were found.
Conclusion
The results of our survey of Wolbachia infection prevalence and genotype diversity in Palearctic D. melanogaster populations confirm previous studies. Wolbachia is ubiquitous in the Palearctic region. The wMel genotype is dominant with local occurrence of rare genotypes. Together with variants of the V mtDNA clade, the variants of the ‘III+’ clade are dominant in both infected and uninfected flies of Palearctic populations. Based on our data on Wolbachia and mtDNA in different years in some Palearctic localities, we can conclude that flies that survive the winter make the predominant symbiont contribution to the subsequent generation. A comprehensive overview of mtDNA and Wolbachia infection of D. melanogaster populations worldwide does not support the recent global Wolbachia genotype replacement hypothesis. However, we cannot exclude wMelCS genotype rate fluctuations in the past.
Similar content being viewed by others
Background
Bacteria of the Wolbachia genus are widespread across Drosophila species [1, 2]. Drosophila spp. are popular model organisms for studies of different aspects of Wolbachia biology including microevolution and population dynamics [2,3,4,5,6,7,8]. Wolbachia can affect the Drosophila host in different ways including male killing in D. bifasciata and D. innubila [9, 10], cytoplasmic incompatibility (CI) in D. paulistorum, D. simulans and D. melanogaster [8, 11, 12], protection from RNA-viruses [13, 14], nutrition provisioning [15], fecundity increasing [16] and suppression of mutations [17,18,19].
Many Wolbachia strains in different Drosophila hosts are non-related. There are, five genetically distant Wolbachia strains in D. simulans, which differ in geographical distribution and CI expression [1, 20, 21]. These strains are coinherited with three mtDNA haplotypes (siI, siII and siIII) [1, 21, 22], which suggests that at least some of these Wolbachia strains have been recently harboured by D. simulans [2].
In D. melanogaster, the single Wolbachia strain, wMel, has been described based on the analysis of some housekeeping genes [23,24,25,26]. Studies of Wolbachia genomes in different D. melanogaster strains have revealed differences in chromosomal rearrangements, variation in indels and repetitive sequences [27,28,29,30,31]. These findings allow the subdivision of the wMel strain into six closely related variants (namely genotypes). These variants form two groups; MEL includes wMel, wMel2, wMel3 and wMel4 genotypes, and CS includes wMelCS and wMelCS2 [29, 32]. Further, the whole-genome sequence analysis has revealed several clades of the wMel strain [30, 33, 34]. The MEL genotypes correspond to Wolbachia clades I-V and VIII, and the CS genotypes correspond to clade VI [30, 32, 34].
In comparison with D. simulans [21, 22], the number of nucleotide polymorphisms in D. melanogaster mitochondrial DNA is low. Only analysing Wolbachia polymorphisms as genetic marker of maternal inheritance and genome sequence analyses of many D. melanogaster isolates have provided valuable data on D. melanogaster mtDNA variation. D. melanogaster mtDNA and the wMel Wolbachia strain are strictly coinherited. mtDNA lineages have been designated as M- or S-clade according to polymorphisms of some loci [32, 35], and as clades I-VI and VIII according to whole-genome sequencing [30, 33, 34]. As a result, the following associations are observed: Wolbachia of MEL group/I-V and VIII clades are associated with mtDNA of M/I-V and VIII clades, and Wolbachia of the CS group/VI clade are associated with mtDNA of the S/VI clade. Phylogenetic analyses revealed the divergence of the wMel Wolbachia strain and D. melanogaster mtDNA from a common ancestor several thousand years ago [30, 32,33,34].
There is a global geographic pattern of D. melanogaster maternal lineages. The MEL-group genotypes and M-clade mtDNA of uninfected flies have been found in D. melanogaster populations all over the world [29, 32, 36, 37]. However, this Wolbachia group and mtDNA includes lineages of I-V and VIII clades (both mtDNA and Wolbachia) that have different geographical distributions. The I and III clades of mtDNA and Wolbachia seem to spread across all continents [32,33,34]. The II and IV clades are found in Africa; the V clade in the Palearctic region, and the VIII clade (associated with wMel2 genotype) in Eastern Asia [30, 32, 33, 38]. The VI clade is associated with CS-group Wolbachia. This clade is found all over the world, but its frequency is very low. Previously, an additional mtDNA clade was proposed (clade VII), which is associated with wMelCS2 genotype and distributed in the Palearctic (mainly in Eastern Europe, the Caucasus, Central Asia and South of Western Siberia) [32, 37]. However, Chrostek et al. [30] did not confirm the validity of this clade. Data about diversity and distribution of D. melanogaster mtDNA and Wolbachia from South America and Australia and many territories in Eurasia are lacking.
There are data on how Wolbachia variants of different clades affect D. melanogaster environmental adaptation [30, 38,39,40]. In contrast to D. simulans, Wolbachia in D. melanogaster induce no/low CI or strong CI in the case of ‘young males’ [11, 41, 42]. The reasons for high Wolbachia density in D. melanogaster populations are unclear because the main factors affecting symbiont spreading are weak, and others, such as protection from viral infections and mutation suppression could only have a localised effect.
In the present study, we evaluated Wolbachia and mtDNA prevalence in fly populations across the vast Palearctic region. In particular, we were interested in the frequency and distribution of Wolbachia genotypes and mtDNA variants in Palearctic fly populations. We confirmed previous data on widespread Wolbachia infection, high infection rates and predominance of the wMel Wolbachia genotype in D. melanogaster populations, and show 1) low genetic diversity of CS Wolbachia genotypes, 2) predominance of two mtDNA clades in Palearctic D. melanogaster populations and 3) overwintering flies in urbanized localities. Based on our results, we conclude that global Wolbachia genotype replacement has not occurred in the recent past.
Methods
Sample collection
Our collection includes 1550 D. melanogaster samples from 12 Palearctic regions (43 localities) collected between 1974 and 2015 (Additional file 1). Most of the samples (1505) are from natural populations collected during 2008–2015 and include isofemale lines and alcohol samples. Isofemale lines were analysed in the year of collection to minimize the possibility of stochastic loss of Wolbachia. In addition, 45 laboratory isofemale lines from long-term storage, established between 1974 and 2005, were studied for mtDNA polymorphisms. Some were also studied for genetic diversity of CS Wolbachia genotypes. Samples were examined for i) Wolbachia prevalence and genotype diversity (n = 1251), ii) genetic diversity of CS-genotypes (n = 22 including nine long-term storage mutant stocks); iii) M/S clades of mtDNA (n = 1550. Here 254 samples that were studied for Wolbachia infection in Bykov et al. [37] were included), and iv) I-VIII mtDNA clades (n = 143) (Additional file 2).
Screening and sequencing
DNA extraction was performed according to Ilinsky [32]. Wolbachia symbionts in the collection were analysed by PCR using 81F/691R primers for the wsp gene [43] and 99F/994R for the 16SrRNA gene [44]. Wolbachia genotypes were determined according to Riegler et al. [29]. According to an analysis of nine complete CS Wolbachia genomes from Richardson et al. [33], Chrostek et al. [30] and Versache et al. [38], we found 30 SNP sites among the CS genotypes (Additional file 3). Nineteen sites were non-parsimonious or uncertain and eleven were parsimonious. We chose three parsimonious sites located in coding regions and separated by more than 89 kbp (Additional file 3) to characterize 22 Wolbachia isolates with primers WclpBF: 5′-GGCTTTCGCAAGTTCGGTTT-3′, WclpBR: 5′-GGAGAGCTGATGTATGGTGT-3′ (208019–208326 region in Wolbachia genome according to GenBank AE017196.1), WlonF: 5′-CAAGTGATGATCCGTAAAGT-3′, WlonR: 5′-GGCATAGAGAAAGTAAAAAGA-3′ (297780–298135 region), WmaeBF: 5′-CTGTGTGATAAGCAAGGAGT-3′, WmaeBR: 5′-TGGGTCAAATGGAGTAGGTA-3′ (469653–470116 region).
All samples were analysed for 2187C/T variants of D. melanogaster mtDNA by PCR with specific primers [32]. These variants correspond to the most ancient split in evolution of D. melanogaster mtDNA. In other words, they are markers of M- and S-clades [32]. The 343 bp mtDNA region (4586–4928 GenBank NC001709) of 143 samples was amplified with primers 04 and At6R [32] and sequenced to determine mitochondrial clades. Polymorphisms in this region allow the identification of III-, V-, VI-, I_II_III- and IV_VIII clades of mtDNA (Additional file 2). Samples of the M-mitotype (n = 111) were randomly chosen from both infected and uninfected flies with the addition of two samples harbouring wMel2. All available S-mitotype samples (n = 32) were analysed.
Amplicons were purified using a Zymoclean™ Gel DNA Recovery Kit (Zymo Research, USA) according to the manufacturer’s instructions, and sequenced by BigDye® Terminator v3.1 cycle sequencing kit (Applied Biosystems). Sequences were deposited in GenBank under accession numbers MG197842 – MG197984 for mtDNA analysis, and MG241453 – MG241491, MH010806 – MH010832 (Additional file 4).
All statistic calculations were performed in MS Excel (Microsoft Corporation) with the AtteStat 12.0.5 add-in.
Results
Wolbachia prevalence in Palearctic populations of D. melanogaster
To estimate Wolbachia prevalence, the 1251 D. melanogaster samples were examined. Wolbachia were found in all studied D. melanogaster Palearctic populations in the range of 0.33–1.0, with an average of 0.56 (Table 1; Additional file 5). The largest numbers of samples were collected from Kaliningrad, Crimea, Sakhalin localities and Nalchik city. In Kaliningrad Oblast, Wolbachia prevalence did not differ significantly (Fisher’s exact test, P = 0.06) over a two-year sampling period. There were also no differences in populations over four years in Nalchik including data from Bykov et al. [37] (Pearson’s chi-square, P = 0.35), or over a two-year period in Sakhalin (Fisher’s exact test, P = 0.82). The population of Izobilnoe (Crimea), which lives on the grape seed dump of the winery industry, had the greatest density of flies. The grape seed piles were swarmed with flies, and we assumed the population numbered at least hundreds of thousands of D. melanogaster individuals within a limited area. Wolbachia prevalence in this population was not different from the average prevalence rate of Palearctic populations (Fisher’s exact test, P = 0.55).
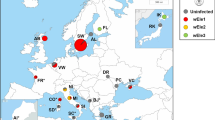
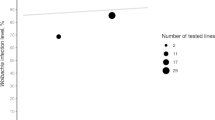
Prevalence rates were compared in relation to latitude and longitude. Our data contained a latitude gap of 30°N-40°N and a longitude gap of 90°E-130°E. Therefore, the population locality positions of ~ 41°N-57°N and ~ 3°E-87°E were considered. As expected, Palearctic populations did not show any geographical pattern of Wolbachia prevalence (Fig. 1, Additional file 6), which is consistent with Kriesner et al. [45] but based on a larger sample size.

Wolbachia frequencies and genotype distribution in Palearctic populations of D. melanogaster by longitude (a) and latitude (b). For more information, see Additional file 6
Wolbachia genetic diversity in the populations
Five of six reported Wolbachia genotypes were found in Palearctic D. melanogaster populations (Table 1, Fig. 1). wMel was the only genotype in the majority of European localities and was predominant in other regions. It is noteworthy that the well-sampled Kaliningrad Oblast region had no cases of non-wMel genotypes, whereas other regions with a large sample size and some regions with small samples contained non-wMel variants, albeit at low frequencies. The high frequency of the wMel4 genotype detected in Sharm el-Sheikh may be explained by genetic drift in the small population. We also found two strains harbouring Wolbachia of the wMel2 genotype in one of two Far East localities. Two cases of wMelCS were found in the south of Western Siberia and Central Asia, wMelCS2 was found in the North Caucasus [37] and two localities of Crimea (Table 1, Fig. 1). Hence, our result confirms and expands upon previously reported data on Palearctic D. melanogaster populations [36, 46].
CS genotype variation
Wolbachia variants of the CS group are rare in field collections but broadly distributed globally. Their genomes are very similar [30, 33, 38]. We checked for genetic differences among CS isolates of various origins. Three Wolbachia genome regions with parsimonious sites for every wMelCS and wMelCS2 sample were sequenced. Only two haplotypes were found (Table 2, Additional file 4), ‘CCG’ was common among wild-type and mutant stocks, and ‘CTG’ was found in only three samples of mutant stocks harbouring the wMelCS genotype. Therefore, we conclude that there is low genetic variation in Wolbachia of the CS group, which appears to contradict the idea of domination such variants in the past [29].
mtDNA variants in the populations
Samples of mtDNA from infected and uninfected D. melanogaster populations were tested for 2187C/T variants. The purpose of this analysis was i) to check any facts of mtDNA/Wolbachia coinheritance disorders, which would indicate horizontal transmission of Wolbachia or mtDNA paternal passing, and ii) to compare M/S ratios in infected vs. uninfected flies, which would indirectly indicate the origin of uninfected flies.
No mtDNA/Wolbachia coinheritance disorders were revealed. All samples infected with Wolbachia of the MEL genotype were M-mitotype, and all samples from the CS group were S-mitotype. In uninfected samples, the M-mitotype was found in all localities, whereas the S-mitotype was only found in North Caucasus and the Far East (Table 1). No significant difference was revealed in M/S ratios of infected vs. uninfected lines (Fisher’s exact test, P = 0.114), which statistically indicates the recent loss of the infection by ancestors of S-mitotype uninfected flies. These results confirm the previous data of Ilinsky [32].
To characterize the mitotype diversity in detail, the 343 bp mtDNA region was sequenced for 143 samples. Among M-mitotype samples of the I_II_III group, III and V clades were found in similar proportion across the studied Palearctic territory (Table 3). The proportion, in particular, I_II_III group + III clade (‘III+’ clade) vs. V clade was not significantly different (Fisher’s exact; P = 0.55). There was no difference in infection prevalence in ‘III+’ vs. the V clade (Fisher’s exact; P = 1.0). Two samples with IV_VIII clades (both wMel2-infected), were found only in the Far East population. All S-mitotype samples from Eastern Europe to the Far East were confirmed to have the canonical VI-clade sequence.
Discussion
Here, the mtDNA and Wolbachia endosymbiont of D. melanogaster were examined in population and phylogeographic terms across the vast Palearctic territory. Our data are consistent with previous observations on i) widespread Wolbachia infection in D. melanogaster populations [29, 35, 47], ii) predominance of the wMel genotype [29, 35], and iii) strict coinheritance of Wolbachia and mtDNA variants [32,33,34, 36, 48]. We demonstrate that fly populations of the temperate zone renew after a cold season. No changes were observed in diversity and rate of maternal factors in D. melanogaster populations of Central (Kaliningrad) and Eastern Europe (Nalchik), and Western Siberia (Altai). No geographical pattern of Wolbachia infection rate was observed in the Palearctic, which corresponds well with the results of Kriesner et al. [45]. However, rare Wolbachia variants were found in certain regions, viz. wMelCS2 from Western Siberia to Eastern Europe [29, 35, 36], wMel2 in the Far East: China, Japan [30, 34], and Sakhalin; wMel4 in Sinai peninsula [32].
In the present survey, Wolbachia variants of the CS group were identified. The wMelCS2 genotype was detected in Eastern Europe and North Caucasus, and two samples with wMelCS infection were found in Western Siberia and Central Asia. Riegler et al. [29], proposed a hypothesis of wMelCS replacement by wMel in the XX century based on the observation that wMelCS is primarily present in populations before 1970 and further the wMel genotype become dominant. If that is the case, high genetic diversity for CS Wolbachia group and S-clade mitochondrial DNA should be observed, whereas diversity of the MEL group and M-clade mtDNA should be rather low. In fact, we observed the opposite situation. There were several lineages within the MEL group and M-clade mtDNA, and only one lineage for the CS group and S-clade mtDNA [30, 33]. Here, we tried to reveal genetic differentiation of CS genotypes using three SNPs located in different protein-coding genes. It is obvious the ‘CCG’ haplotype is an ancestral, as it is shared by both MEL and CS Wolbachia groups. Moreover, the ‘CCG’ haplotype was found among wMelCS and wMelCS2 genotypes, and other haplotypes seem to be local variants, namely ‘TCG’ in North America and ‘CCT’ in South-East Asia. Several isolates of the ‘CTG’ haplotype, which are found in wMelCS-infected stocks, could be the result of using one or more sources of maternal laboratory stock(s). Thus, we observed low genetic variation within the CS lineage of Wolbachia, which, together with above-mentioned comparison of MEL/M and CS/S diversity, and low diversity of the VI mtDNA lineage contradicts the hypothesis of recent replacement of Wolbachia genotypes. It is difficult to imagine different mitotypes supplanting the VI clade variants. The reason for the high proportion of wMelCS genotypes in populations before 1970 could be a case of sample error. Indeed, the number of stocks established with flies collected before the 1980s is very low (Additional file 5). This inference should be confirmed by more detailed analyses of both Wolbachia and mtDNA. An alternative scenario that cannot be ruled out is an increase in the wMelCS genotype rate in D. melanogaster populations during the first part of the XX century or earlier. This increasing could be due to specific interactions between wMelCS Wolbachia genotypes with unknown factors, the latter could be sigma virus [49, 50] or P-element [51, 52] that have recently invade D. melanogaster populations.
Coinheritance of Wolbachia variants and host mtDNA haplotypes has been reported for different species [1, 7, 21, 53,54,55,56,57,58]. The association between D. melanogaster mtDNA and Wolbachia genotypes has also been demonstrated in several studies [32,33,34]. Our M and S mtDNA clades distribution data in Palearctic D. melanogaster populations and their coinheritance with Wolbachia genotypes confirms the strict association between symbiont and mitochondrial lineages. Two main D. melanogaster mtDNA lineages (clades) within the M-clade were revealed. The ‘III+’ lineage consists of clades I and III, which are widespread all over the world [30, 32,33,34, 38], and clade II, which was found only in African D. melanogaster populations [33, 34]. Some samples of the ‘III+’ clade were identified as clade III by 4616A/T (Additional file 1). Therefore, we cannot exclude that samples with the 4616(A) substitution could also be clade III, and further analysis is required. The V clade of mtDNA was previously reported in D. melanogaster populations from Western Europe [33, 34, 38], Central and North Asia [32]. In the present study, this clade was also found in the Far East. Thus, we can assume that the V mtDNA clade is common for Palearctic D. melanogaster populations. D. melanogaster with the V clade of mtDNA were previously shown to be more viable in cold conditions than other clades [38], that could explain high frequency of this clade in Palearctic. However, the question of a cold tolerance mechanism determined by mtDNA remains unclear. The same rate of Wolbachia infection within D. melanogaster with ‘III+’ and V mtDNA clades may indicate a recent loss of the symbiont.
Conclusions
Our in-depth survey of maternal-inherited factors of Palearctic D. melanogaster populations is consistent with previous studies and expands our knowledge. Prevalence of Wolbachia infection does not have a specific distribution pattern in the Palearctic. The wMel genotype inhabits every population, whereas other genotypes are rare and localised. Variants of V and ‘III+’ mitochondrial clades predominate in infected and uninfected flies across the Palearctic territory. According to symbiont and mtDNA diversity, the fly populations of many regions in temperate zones renew after the cold season, and the contribution of fly migration is not detected. Low genetic polymorphism of CS genotypes together with mitotypes and Wolbachia infection of global D. melanogaster populations do not support the hypothesis of a recent global Wolbachia genotype replacement. However, an increase in the wMelCS genotype rate in global D. melanogaster populations due to interactions with specific factors cannot be excluded.
References
Mercot H, Charlat S. Wolbachia infections in Drosophila melanogaster and D. simulans: polymorphism and levels of cytoplasmic incompatibility. Genetica. 2004;120:51–9.
Turelli M, Cooper BS, Richardson KM, Ginsberg PS, Peckenpaugh B, Antelope CX, et al. Rapid global spread of wRi-like Wolbachia across multiple Drosophila. Curr Biol. 2018;28(6):963–71.
Hoffmann AA, Turelli M, Harshman LG. Factors affecting the distribution of cytoplasmic incompatibility in Drosophila simulans. Genetics. 1990;126(4):933–48.
Hoffmann AA, Clancy DJ, Merton E. Cytoplasmic incompatibility in Australian populations of Drosophila melanogaster. Genetics. 1994;136(3):993–9.
Turelli M, Hoffmann AA, McKechnie SW. Dynamics of cytoplasmic incompatibility and mtDNA variation in natural Drosophila simulans populations. Genetics. 1992;132(3):713–23.
Turelli M, Hoffmann AA. Cytoplasmic incompatibility in Drosophila simulans: dynamics and parameter estimates from natural populations. Genetics. 1995;140(4):1319–38.
Shoemaker DD, Dyer KA, Ahrens M, McAbee K, Jaenike J. Decreased diversity but increased substitution rate in host mtDNA as a consequence of Wolbachia endosymbiont infection. Genetics. 2004;168(4):2049–58.
Miller WJ, Ehrman L, Schneider D. Infectious speciation revisited: impact of symbiont-depletion on female fitness and mating behavior of Drosophila paulistorum. PLoS Pathog. 2010;6(12):e1001214.
Veneti Z, Toda MJ, Hurst GDD. Host resistance does not explain variation in incidence of male-killing bacteria in Drosophila bifasciata. BMC Evol Biol. 2004;4:52.
Dyer KA, Minhas MS, Jaenike J. Expression and modulation of embryonic male-killing in Drosophila innubila: opportunities for multilevel selection. Evolution. 2005;59(4):838–48.
Hoffmann AA. Partial cytoplasmic incompatibility between 2 Australian populations of Drosophila melanogaster. Entomol Exp Appl. 1988;48:61–7.
Hoffmann AA, Turelli M, Simmons GM. Unidirectional incompatibility between populations of Drosophila simulans. Evolution. 1986;40:692–701.
Hedges LM, Brownlie JC, O'neill SL, Johnson KN. Wolbachia and virus protection in insects. Science. 2008;322(5902):702.
Teixeira L, Ferreira Á, Ashburner M. The bacterial symbiont Wolbachia induces resistance to RNA viral infections in Drosophila melanogaster. PLoS Biol. 2008;6(12):e1000002.
Brownlie JC, Cass BN, Riegler M, Witsenburg JJ, Iturbe-Ormaetxe I, McGraw EA, O’Neill SL. Evidence for metabolic provisioning by a common invertebrate endosymbiont, Wolbachia pipientis, during periods of nutritional stress. PLoS Pathog. 2009;5:e1000368.
Weeks AR, Turelli M, Harcombe WR, Reynolds KT, Hoffmann AA. From parasite to mutualist: rapid evolution of Wolbachia in natural populations of Drosophila. PLoS Biol. 2007;5:e114.
Starr DJ, Cline TW. A host parasite interaction rescues Drosophila oogenesis defects. Nature. 2002;418:76–9.
Clark ME, Anderson CL, Cande J, Karr TL. Widespread prevalence of Wolbachia in laboratory stocks and the implications for Drosophila research. Genetics. 2005;170:1667–75.
Ikeya T, Broughton S, Alic N, Grandison R, Partridge L. The endosymbiont Wolbachia increases insulin/IGF-like signalling in Drosophila. Proc R Soc B. 2009;276(1674):3799–807.
Turelli M, Hoffmann AA. Rapid spread of an inherited incompatibility factor in California Drosophila. Nature. 1991;353(6343):440.
Rousset F, Solignac M. Evolution of single and double Wolbachia symbioses during speciation in the Drosophila simulans complex. Proc Natl Acad Sci U S A. 1995;92:6389–93.
Baba-Aïssa F, Solignac M, Dennebouy N, David JR. Mitochondrial DNA variability in Drosophila simulans: quasi absence of polymorphism within each of the three cytoplasmic races. Heredity. 1988;61(3):419.
Holden PR, Jones P, Brookfield JF. Evidence for a Wolbachia symbiont in Drosophila melanogaster. Genet Res. 1993;62(1):23–9.
Bourtzis K, Nirgianaki A, Onyango P, Savakis C. A prokaryotic dnaA sequence in Drosophila melanogasten Wolbachia infection and cytoplasmic incompatibility among laboratory strains. Insect Mol Biol. 1994;3(3):131–42.
Werren JH, Zhang W, Guo LR. Evolution and phylogeny of Wolbachia: reproductive parasites of arthropods. Proc R Soc B. 1995;261(1360):55–63.
Zhou W, Rousset F, O'Neill S. Phylogeny and PCR–based classification of Wolbachia strains using wsp gene sequences. Proc R Soc Lond B Biol Sci. 1998;265(1395):509–15.
Sun LV, Foster JM, Tzertzinis G, Ono M, Bandi C, Slatko BE, O'Neill SL. Determination of Wolbachia genome size by pulsed-field gel electrophoresis. J Bacteriol. 2001;183:2219–25.
Sun LV, Riegler M, O'Neill SL. Development of a physical and genetic map of the virulent Wolbachia strain wMelPop. J Bacteriol. 2003;185(24):7077–84.
Riegler M, Sidhu M, Miller WJ, O’Neill SL. Evidence for a global Wolbachia replacement in Drosophila melanogaster. Curr Biol. 2005;15(15):1428–33.
Chrostek E, Marialva MS, Esteves SS, Weinert LA, Martinez J, Jiggins FM, Teixeira L. Wolbachia variants induce differential protection to viruses in Drosophila melanogaster: a phenotypic and phylogenomic analysis. PLoS Genet. 2013;9(12):e1003896.
Chrostek E, Teixeira L. Mutualism breakdown by amplification of Wolbachia genes. PLoS Biol. 2015;13(2):e1002065.
Ilinsky Y. Coevolution of Drosophila melanogaster mtDNA and Wolbachia genotypes. PLoS One. 2013;8(1):e54373.
Richardson MF, Weinert LA, Welch JJ, Linheiro RS, Magwire MM, Jiggins FM, Bergman CM. Population genomics of the Wolbachia endosymbiont in Drosophila melanogaster. PLoS Genet. 2012;8(12):e1003129.
Early AM, Clark AG. Monophyly of Wolbachia pipientis genomes within Drosophila melanogaster: geographic structuring, titre variation and host effects across five populations. Mol Ecol. 2013;22(23):5765–78.
Ilinsky YY, Zakharov IK. Infection of the Uman’ population of Drosophila melanogaster with the cytoplasmic endosymbiont Wolbachia. Dokl Biol Sci MAIK Nauka/Interperiodica. 2007a;413(1):166–8.
Ilinsky YY, Zakharov IK. The endosymbiont Wolbachia in Eurasian populations of Drosophila melanogaster. Russ J Genet. 2007b;43(7):748–56.
Bykov RA, Ilinskii YY, Voloshina MA, Zakharov IK. Prevalence and genotypic diversity of the symbiotic bacterium Wolbachia in the Drosophila melanogaster population of Nalchik. Russ J Genet. 2014;4(6):539–42.
Versace E, Nolte V, Pandey RV, Tobler R, Schlötterer C. Experimental evolution reveals habitat-specific fitness dynamics among Wolbachia clades in Drosophila melanogaster. Mol Ecol. 2014;23(4):802–14.
Faria VG, Martins NE, Magalhães S, Paulo TF, Nolte V, Schlötterer C, Sucena E, Teixeira L. Drosophila adaptation to viral infection through defensive symbiont evolution. PLoS Genet. 2016;12(9):e1006297.
Gruntenko NЕ, Ilinsky YY, Adonyeva NV, Burdina EV, Bykov RA, Menshanov PN, Rauschenbach IY. Various Wolbachia genotypes differently influence host Drosophila dopamine metabolism and survival under heat stress conditions. BMC Evol Biol. 2017;17(2):252.
Yamada R, Floate KD, Riegler M, O'Neill SL. Male development time influences the strength of Wolbachia-induced cytoplasmic incompatibility expression in Drosophila melanogaster. Genetics. 2007;177(2):801–8.
Ilinsky YY, Zakharov IK. Cytoplasmic incompatibility in Drosophila melanogaster is caused by different Wolbachia genotypes. Russ J Genet. 2011;1(5):458.
Braig HR, Zhou W, Dobson SL, O’Neill SL. Cloning and characterization of a gene encoding the major surface protein of the bacterial endosymbiont Wolbachia pipientis. J Bacteriol. 1998;180(9):2373–8.
O'Neill SL, Giordano R, Colbert AM, Karr TL, Robertson HM. 16S rRNA phylogenetic analysis of the bacterial endosymbionts associated with cytoplasmic incompatibility in insects. Proc Natl Acad Sci. 1992;89(7):2699–702.
Kriesner P, Conner WR, Weeks AR, Turelli M, Hoffmann AA. Persistence of a Wolbachia infection frequency cline in Drosophila melanogaster and the possible role of reproductive dormancy. Evolution. 2016;70(5):979–97.
Serga S, Maistrenko O, Rozhok A, Mousseau T, Kozeretska I. Fecundity as one of possible factors contributing to the dominance of the wMel genotype of Wolbachia in natural populations of Drosophila melanogaster. Symbiosis. 2014;63(1):11–7.
Solignac M, Vautrin D, Rousset F. Widespread occurence of the proteobacteria Wolbachia and partial cytoplasmic incompatibility in Drosophila melanogaster. Comptes rendus de l'Académie des sciences. Série 3, Sciences de la vie. 1994;317(5):461–70.
Nunes MD, Nolte V, Schlötterer C. Nonrandom Wolbachia infection status of Drosophila melanogaster strains with different mtDNA haplotypes. Mol Biol Evol. 2008;25(11):2493–8.
Fleuriet A. Perpetuation of the hereditary sigma virus in populations of its host, Drosophila melanogaster. Geographical analysis of correlated polymorphisms. Genetica. 1986;70(3):167–77.
Carpenter JA, Obbard DJ, Maside X, Jiggins FM. The recent spread of a vertically transmitted virus through populations of Drosophila melanogaster. Mol Ecol. 2007;16(18):3947–54.
Anxolabéhère D, Kidwell MG, Periquet G. Molecular characteristics of diverse populations are consistent with the hypothesis of a recent invasion of Drosophila melanogaster by mobile P elements. Mol Biol Evol. 1988;5(3):252–69.
Engels WR. Invasions of P elements. Genetics. 1997;145(1):11.
Marcade I, Souty-Grosset C, Bouchon D, Rigaud T, Raimond R. Mitochondrial DNA variability and Wolbachia infection in two sibling woodlice species. Heredity. 1999;83(1):71.
Rokas A, Atkinson RJ, Brown GS, West SA, Stone GN. Understanding patterns of genetic diversity in the oak gallwasp Biorhiza pallida: demographic history or a Wolbachia selective sweep? Heredity. 2001;87(3):294.
Hinrich J, Vd Schulenburg G, Hurst GD, Tetzlaff D, Booth GE, Zakharov IA, Majerus ME. History of infection with different male-killing bacteria in the two-spot ladybird beetle Adalia bipunctata revealed through mitochondrial DNA sequence analysis. Genetics. 2002;160(3):1075–86.
Rasgon JL, Cornel AJ, Scott TW. Evolutionary history of a mosquito endosymbiont revealed through mitochondrial hitchhiking. Proc R Soc B. 2006;273(1594):1603–11.
Dumas E, Atyame CM, Milesi P, Fonseca DM, Shaikevich EV, Unal S, Makoundou P, Weill M, Duron O. Population structure of Wolbachia and cytoplasmic introgression in a complex of mosquito species. BMC Evol Biol. 2013;13(1):1.
Chen F, Coates B, He KL, Bai SX, Zhang TT, Wang ZY. Effects of Wolbachia on mitochondrial DNA variation in populations of Athetis lepigone (Lepidoptera: Noctuidae) in China. Mitochondrial DNA Part A. 2017;28(6):826–34.
Acknowledgements
The authors thank Andrey Broshkov for technical support; Prof. Edward Dubrovsky and Veronika Dubrovskaya for critically reading of the manuscript.
Funding
This study was supported by the RFBR No 16–34-00819, RFBR No 16–04-00980, the State Budgeted Project No 0324-2019-0041 and partially by the RFBR No 16–04-00060.
Publication costs are funded by Immanuel Kant Baltic Federal University.
Availability of data and materials
The datasets supporting the conclusions of this article are included in this published article and its supplementary information files. The sequence data used in this study have been submitted to the GenBank databases under accession numbers: MG197842-MG197984, MG241453-MG241491, MH010806-MH010832.
About this supplement
This article has been published as part of BMC Evolutionary Biology Volume 19 Supplement 1, 2019: Selected articles from BGRS\SB-2018: evolutionary biology. The full contents of the supplement are available online at https://bmcevolbiol.biomedcentral.com/articles/supplements/volume-19-supplement-1.
Author information
Authors and Affiliations
Contributions
RAB and YYI conceived and designed the study. RAB, MAY and ESM performed experimental procedures and analyses. RAB, YYI, MVD, ESM and MAV collected flies in the field. IKZ provided laboratory stocks. IOM and MVD provided reagents, materials and analysis tools. RAB, YYI and NEG drafted the manuscript. NEG also provided support and discussions. All authors gave their approval for this publication.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
List of Drosophila melanogaster stocks used for analysis of 343 bp mtDNA region. GenBank numbers of sequencing samples are indicated. (XLS 55 kb)
Additional file 2:
Polymorphic sites of 343 bp mtDNA region of Drosophila melanogaster (according to GenBank NC001709). (XLS 37 kb)
Additional file 3:
Haplotypes of Wolbachia from CS-group genotypes. GenBank accession numbers are indicated in brackets where available. (DOC 74 kb)
Additional file 4:
Nucleotide polymorphisms of CS Wolbachia variants. Positions chosen for analysis performed in the current study are highlighted in yellow. Uncertain nucleotides highlighted in grey. (XLS 43 kb)
Additional file 5:
Dataset of Wolbachia genotypes found in worldwide populations of Drosophila melanogaster during the 1925–2015 period. (XLS 72 kb)
Additional file 6:
Dataset used for analysis of geographical Wolbachia patterns. (XLS 31 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bykov, R.А., Yudina, M.A., Gruntenko, N.E. et al. Prevalence and genetic diversity of Wolbachia endosymbiont and mtDNA in Palearctic populations of Drosophila melanogaster. BMC Evol Biol 19 (Suppl 1), 48 (2019). https://doi.org/10.1186/s12862-019-1372-9
Published:
DOI: https://doi.org/10.1186/s12862-019-1372-9