
Abstract
Background
With advancements in high-throughput technologies, the cost of obtaining expression profiles of both mRNA and microRNA in the same individual has substantially decreased. Integrated analysis of these profiles can help to elucidate the functional effects of RNA expression in complex diseases, such as cancer. However, fundamental discrepancies are observed in the results from microRNA-mRNA target gene prediction algorithms, and few packages can be used to analyze microRNA and mRNA expression levels simultaneously.
Results
To address these issues, an R package, anamiR, was developed. A total of 10 experimental/prediction databases were integrated. Two analytical functions are provided in anamiR, including the single marker test and functional gene set enrichment analysis, and several parameters can be changed by users. Here we demonstrate the potential application of the anamiR package to 2 publicly available microarray datasets.
Conclusion
The anamiR package is effective for an integrated analysis of both RNA and microRNA profiles. By characterizing biological functions and signaling pathways, this package helps identify dysregulated genes/miRNAs from biological and medical experiments. The source code and manual of the anamiR package are freely available at https://bioconductor.org/packages/release/bioc/html/anamiR.html.
Similar content being viewed by others


Background
With advancements in high-throughput technologies, the cost of analyzing multiple gene expression profiles in the same individual has dropped substantially. Many studies have described attempts to analyze RNA and microRNA (miRNA) profiles simultaneously. To date, only a few miRNAs and their target genes have been validated through biological experiments [1], and thus prediction algorithms have been widely used to identify potential miRNA-gene interaction pairs. The most popular method for predicting target genes of miRNAs is that of matching the 3’UTR of an mRNA to the “seed region” (a conserved sequence of 2 to 8 nucleotides) of an miRNA, which assumes perfect Watson-Crick complementarity between the mRNA 3’UTR and the miRNA. In addition, the seed region can be used to classify families and species of miRNAs. Consequently, the seed region has been demonstrated to be a key element of miRNA-target gene prediction [2]. However, other research has shown that the pairing mechanism between miRNA and mRNA can occur anywhere along the entire mRNA, which suggests that algorithms focusing on the seed regions can only identify a subset of all potential miRNA-target gene pairs [3]. To address this issue, other popular computational approaches, such as free energy minimization and machine learning, have been developed. Measuring the minimum free energy can help to assess the stability of binding sites between miRNA and target genes. A predicted miRNA-target pair with lower free energy indicates that the binding is more stable and thus more likely to be a true result [2, 4]. With the rapid accumulation of massive amounts of data, machine learning algorithms have also been implemented in many prediction algorithms through training processes containing the entire dataset of known miRNA targets [4]. In such approaches, important features that can facilitate identification of possible target genes from miRNAs can be revealed. Good performance has been reported for support vector machine [5] and generalized linear model [6] algorithms. Furthermore, two machine learning based algorithms were implemented to dissect the associations between miRNAs and diseases [7, 8], and their results demonstrated that utilizing data mining approaches may effectively improve the prediction accuracy. However, prediction results from different algorithms usually show large discrepancies [9]. For example, the proportion of the miRNA-gene pairs that can be predicted by all 8 algorithms in our system is only 0.001% (Additional file 1: Table S1). Therefore, inconsistent prediction results still pose a major challenge to advanced analyses of miRNA targets. To address this issue, one possible solution is to use several algorithms to analyze several experimental datasets at the same time [10]. In this study, we developed an R package, anamiR, for analyzing miRNA expression and gene expression concurrently. A total of 8 target prediction algorithms and 2 experimentally validated miRNA databases were included. Users can analyze genome-wide expression profiles of both miRNA and RNA without applying a preset filter. The anamiR package provides 2 major functions: the single marker test and functional gene set enrichment analysis. The former can be used to identify differentially expressed RNAs/miRNAs, and the latter is used to characterize their biological functions and signaling pathways. Alternatively, users can pre-select gene sets and/or pathways of interest, and the anamiR package can reveal dysregulated genes/miRNAs involved in them. The anamiR package can substantially reduce the time and effort required to perform an integrated analysis of genome-wide miRNA and gene expression.
Implementation
Characteristics of anamiR
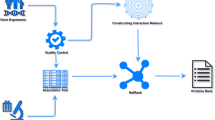
The overall structure of the anamiR package is illustrated in Fig. 1. We collected both predicted and validated datasets containing the potential target genes of miRNAs, as well as biological functions and pathway information, in the anamiR database. To address the issue of low consistency across independent prediction algorithms, we utilized an approach that selects the intersection of the prediction results. The same approach has been used in our previous study [10]. Compared with the previous study, we have added 8 new or updated prediction algorithms, including different in silico approaches for predicting potential miRNA-gene pairs, such as strong base pairing between 3′ UTR of mRNAs and seed regions with variously complementary types (i.e., TargetScan [11] and EIMMo [12]), additionally considering thermal dynamic stability of binding sites (i.e., rna22 [13], miRanda [14], MicroCosm [15], and PITA [16]), incorporating machine learning methods (i.e., DIANA-microT-CDS [17] and miRDB [5]), and 2 experimentally validated datasets (i.e., miRecords [18], and miRTarBase [19]) in anamiR. The numbers of miRNA-gene interaction pairs are summarized in Additional file 1: Table S2. Also, four datasets containing information on biological functions and pathways in both humans and mice were embedded in anamiR, including KEGG [20], Reactome [21], BioCarta, and MouseCyc [22]. Currently, only one published R package, miRComb [23], has similar function to anamiR, and comparisons of the two are shown in Additional file 1: Table S3. The primary advantages of anamiR with respect to miRComb are the inclusion of experimentally validated datasets and the greater number of functional prediction algorithms.

An overview of anamiR. The rounded rectangle with a dotted outline shown in grey indicates the core of the anamiR package. The input data and output results are shown in green. The blue boxes represent two major workflows in the package. Three databases including pathway information from four datasets (orange boxes), collections of miRNA- gene interaction pairs from eight prediction programs (white boxes), and two experimentally validated datasets (purple boxes), and MSigDB are shown in yellow cylinders
Analytical workflows in the anamiR package
In the anamiR package, we have provided a general workflow that contains several statistical tests for performing differential expression analysis, correlation analysis, and functional analysis (Fig. 2a). Users need to provide 2 data sheets of genome-wide expression levels of both miRNA and genes, and information on sample grouping is also required to perform the statistical analyses. For each statistical test and analysis step, all results can be generated as output, and most analysis parameters are flexible and can be changed by the user. To estimate the chances of randomly identifying a significantly enriched biological function, a permutation test is performed.

The proposed workflows in the anamiR package. (a) The general workflow provides six steps including normalization (optional), differential expression analysis, conversion of miRNA names (optional), correlation analysis, database intersection, and functional analysis. miRNA-gene interaction pairs and correspondingly enriched pathways can be identified from the workflow using both mRNA and miRNA expression data. (b) The function-driven analysis workflow is performed to identify significantly dysregulated pathways and to obtain potential miRNA-gene interaction pairs using genome-wide expression profiles. Each box shown in blue represents a function in the anamiR package. The input data and output results are shown in green. Yellow cylinders indicate databases for query and numbers of dataset collections are shown in parentheses
In addition to the general workflow, the anamiR package provides another workflow called function-driven analysis (Fig. 2b). Users can select the biological functions/pathways of interest on which to perform these integrated analyses. All genes in the corresponding functional pathways are analyzed, with no filtering based on their statistical P-values. Such an approach can not only reduce the number of tests required to obtain an answer, but also take moderate gene expression changes into consideration, even if they do not reach statistical significance.
Statistical approaches
In the anamiR package, we have provided four statistical tests and/or algorithms by which to identify differentially expressed genes and/or miRNAs: limma [24], t-test, Wilcoxon rank sum test, and DESeq2 [25]. The default P-value threshold for the statistical tests is defined as 0.05 and the default threshold for the expression ratio between two phenotypes is set at 0.5. Both parameters are user-adjustable. The Benjamini-Hochberg method is performed to address the multiple testing issue. Three measurements of the trend in the expression levels between gene and miRNA are provided: Pearson correlation coefficient, Kendall rank correlation coefficient, and Spearman’s rank correlation coefficient. Since the expression level of a miRNA is negatively correlated to the expression level of its target gene, the default cutoff of the correlation (also user-adjustable) is defined as − 0.5, which corresponds to moderate correlation (Additional file 1: Table S4 and Table S5). For those genes showing negative correlation with their regulatory miRNA, a hypergeometric test is performed to identify significantly enriched pathways. Lastly, to address the possibility of identifying a significant pathway through miRNA enrichment analysis by random chance [26], a permutation test is embedded in the anamiR package. The random chance of identifying a significant pathway is obtained by randomly selecting the same number of genes in the enriched pathway and calculating the enriched P-value. This procedure is repeated 5000 times to generate a null distribution, and the empirical P-value is determined by comparing the enrichment P-value obtained from the real data with the P-value from the null distribution. For the function-driven analysis workflow, we employed the gage package [27] to perform genome-wide functional enrichment analysis, and users can manually set the number of functional terms of interest.
Results and discussion
Dataset description
To demonstrate the potential applications of the anamiR package, 2 examples from real microarray datasets are provided below. The first example (GSE16558) has 60 patients with multiple myeloma and 5 normal controls [28], whereas the second example (GSE60371) contains 48 prostate cancer patients and 6 normal controls [29]. Both datasets are composed of paired miRNA and mRNA expression data.
Example 1: multiple myeloma (GSE16558)
We analyzed the GSE16558 dataset by the general workflow algorithm (Fig. 2a), using limma with the default parameters shown in the supplementary information (Additional file 1: Table S6). The 5 interaction pairs with the highest correlation coefficients between miRNA and gene are shown in Table 1. A previous study has identified miR-485-5p as having dysregulated expression levels in multiple myeloma [30], and its target gene, JMY, is reported as a mediating and regulatory protein of TP53 [31]. Intriguingly, 3 out of the top 5 enriched pathways were related to TP53 (Table 2), suggesting the interaction between miR-485-5p and JMY deserves further investigation in multiple myeloma patients.
Alternatively, the top 5 enriched pathways from the function-driven analysis (Fig. 2b) are shown in Table 3. Notably, the most significant pathway was related to ribosome function, and 3 of the 5 interaction pairs with the largest negative correlations include miR-485-5p, in agreement with the results from the general workflow (Additional file 1: Table S7). Therefore, these results demonstrate that anamiR is able to identify important interaction pairs of miRNA and target genes in a specific disease.
Example 2: prostate cancer (GSE60371)
Similar to the previous example, we utilized the general workflow algorithm with the default parameters in GSE60371 (Additional file 1: Table S6). Notably, 2 miRNAs from the miR-320 family were identified (Table 4), and their down-regulation has been reported in prostate cancer [37]. Intriguingly, the overexpression of UBE2C was reported in prostate cancer [38] and its miRNA regulator is miR-320d. Taken together, the dysregulation of the miR-320 family and UBE2C deserve further investigation in prostate cancer.
As shown in Tables 5 and 6, the results showed that the proportion of miRNA-gene pairs showing negative Pearson correlation coefficients increase along with the number of analyzed algorithms, suggesting better prediction performances can be achieved by the integration of multiple algorithms.
Conclusions
The anamiR package provides an integrated approach for identifying paired mRNA and miRNA expression profiles. The general workflow is utilized to predict the target genes and their associated functional pathways for miRNA simultaneously. Within gene sets and pathways of interest, the function-driven analysis workflow is applied to identify miRNA-gene interaction pairs from among the significant gene sets and pathways. We believe that approaches considering the associations between mRNAs and miRNAs, as well as regulation of genes and pathways, can provide insight into dysfunction in cancers.
Availability and requirements
-
Project name: anamiR
-
Project home page: https://bioconductor.org/packages/release/bioc/html/anamiR.html
-
Operating system(s): Platform independent
-
Programming language: R
-
Other requirements: R (> = 3.3.3), SummarizedExperiment (> = 1.1.6), Bioconductor (> = 3.4), stats, DBI, limma, lumi, agricolae, RMySQL, DESeq2, SummarizedExperiment, gplots, gage, S4Vectors
-
License: GNU GPLv2
-
Any restrictions to use by non-academics: None
Abbreviations
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- miRNA:
-
microRNA
References
Kuhn DE, Martin MM, Feldman DS, Terry AV, Jr., Nuovo GJ, Elton TS: Experimental validation of miRNA targets. Methods 2008, 44(1):47–54.
Riffo-Campos ÁL, Riquelme I, Brebi-Mieville P. Tools for sequence-based miRNA target prediction: what to choose? Int J Mol Sci. 2016;17(12):1987.
Schnall-Levin M, Rissland OS, Johnston WK, Perrimon N, Bartel DP, Berger B. Unusually effective microRNA targeting within repeat-rich coding regions of mammalian mRNAs. Genome Res. 2011;21(9):1395–403.
Peterson SM, Thompson JA, Ufkin ML, Sathyanarayana P, Liaw L, Congdon CB. Common features of microRNA target prediction tools. Front Genet. 2014;5:23.
Wang X. miRDB: a microRNA target prediction and functional annotation database with a wiki interface. RNA (New York, NY). 2008;14(6):1012–7.
Reczko M, Maragkakis M, Alexiou P, Grosse I, Hatzigeorgiou AG. Functional microRNA targets in protein coding sequences. Bioinformatics. 2012;28(6):771–6.
Jiang L, Ding Y, Tang J, Guo F. MDA-SKF: similarity kernel fusion for accurately discovering miRNA-disease association. Front Genet. 2018;9:618.
Jiang L, Xiao Y, Ding Y, Tang J, Guo F. FKL-Spa-LapRLS: an accurate method for identifying human microRNA-disease association. BMC Genomics. 2018;19(Suppl 10):911.
Zheng H, Fu R, Wang J-T, Liu Q, Chen H, Jiang S-W. Advances in the techniques for the prediction of microRNA targets. Int J Mol Sci. 2013;14(4).
Lu TP, Lee CY, Tsai MH, Chiu YC, Hsiao CK, Lai LC, Chuang EY. miRSystem: an integrated system for characterizing enriched functions and pathways of microRNA targets. PLoS One. 2012;7(8):e42390.
Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20.
Gaidatzis D, van Nimwegen E, Hausser J, Zavolan M. Inference of miRNA targets using evolutionary conservation and pathway analysis. BMC Bioinformatics. 2007;8:69.
Loher P, Rigoutsos I. Interactive exploration of RNA22 microRNA target predictions. Bioinformatics (Oxford, England). 2012;28(24):3322–3.
Betel D, Wilson M, Gabow A, Marks DS, Sander C: The microRNA.org resource: targets and expression. Nucleic Acids Res 2008, 36(Database issue):D149–D153.
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36(Database issue):D154–8.
Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. 2007;39(10):1278–84.
Paraskevopoulou MD, Georgakilas G, Kostoulas N, Vlachos IS, Vergoulis T, Reczko M, Filippidis C, Dalamagas T, Hatzigeorgiou AG. DIANA-microT web server v5.0: service integration into miRNA functional analysis workflows. Nucleic Acids Res. 2013;41(Web Server issue):W169–73.
Xiao F, Zuo Z, Cai G, Kang S, Gao X, Li T. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009;37(Database):D105–10.
Chou CH, Chang NW, Shrestha S, Hsu SD, Lin YL, Lee WH, Yang CD, Hong HC, Wei TY, Tu SJ, et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016;44(D1):D239–47.
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36(Database issue):D480–4.
Croft D, O'Kelly G, Wu G, Haw R, Gillespie M, Matthews L, Caudy M, Garapati P, Gopinath G, Jassal B, et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 2011;39(Database):D691–7.
Evsikov AV, Dolan ME, Genrich MP, Patek E, Bult CJ. MouseCyc: a curated biochemical pathways database for the laboratory mouse. Genome Biol. 2009;10(8):R84.
Vila-Casadesús M, Gironella M, Lozano JJ. MiRComb: an R package to analyse miRNA-mRNA interactions. Examples across five digestive cancers. PLoS One. 2016;11(3):e0151127.
Smyth GK: Limma: linear models for microarray data. In: Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Edited by Gentleman R, Carey V, Dudoit S, R Irizarry WH. New York: Springer; 2005.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Bleazard T, Lamb JA, Griffiths-Jones S. Bias in microRNA functional enrichment analysis. Bioinformatics (Oxford, England). 2015;31(10):1592–8.
Luo W, Friedman MS, Shedden K, Hankenson KD, Woolf PJ. GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics. 2009;10:161.
Gutierrez NC, Sarasquete ME, Misiewicz-Krzeminska I, Delgado M, De Las Rivas J, Ticona FV, Ferminan E, Martin-Jimenez P, Chillon C, Risueno A, et al. Deregulation of microRNA expression in the different genetic subtypes of multiple myeloma and correlation with gene expression profiling. Leukemia. 2010;24(3):629–37.
Dallavalle C, Albino D, Civenni G, Merulla J, Ostano P, Mello-Grand M, Rossi S, Losa M, D'Ambrosio G, Sessa F, et al. MicroRNA-424 impairs ubiquitination to activate STAT3 and promote prostate tumor progression. J Clin Invest. 2016;126(12):4585–602.
Berenstein R, Blau O, Nogai A, Waechter M, Slonova E, Schmidt-Hieber M, Kunitz A, Pezzutto A, Doerken B, Blau IW. Multiple myeloma cells alter the senescence phenotype of bone marrow mesenchymal stromal cells under participation of the DLK1-DIO3 genomic region. BMC Cancer. 2015;15:68.
Rebhan M, Chalifa-Caspi V, Prilusky J, Lancet D. GeneCards: integrating information about genes, proteins and diseases. Trends Genet. 1997;13(4):163.
Raimondi L, De Luca A, Morelli E, Giavaresi G, Tagliaferri P, Tassone P, Amodio N. MicroRNAs: novel crossroads between myeloma cells and the bone marrow microenvironment. Biomed Res Int. 2016;2016:6504593.
Blotta S, Tassone P, Prabhala RH, Tagliaferri P, Cervi D, Amin S, Jakubikova J, Tai YT, Podar K, Mitsiades CS, et al. Identification of novel antigens with induced immune response in monoclonal gammopathy of undetermined significance. Blood. 2009;114(15):3276–84.
Liu Z, Zhang G, Yu W, Gao N, Peng J. miR-186 inhibits cell proliferation in multiple myeloma by repressing Jagged1. Biochem Biophys Res Commun. 2016;469(3):692–7.
Xu H, Liu C, Zhang Y, Guo X, Liu Z, Luo Z, Chang Y, Liu S, Sun Z, Wang X. Let-7b-5p regulates proliferation and apoptosis in multiple myeloma by targeting IGF1R. Acta Biochim Biophys Sin Shanghai. 2014;46(11):965–72.
Rossi M, Tagliaferri P, Tassone P. MicroRNAs in multiple myeloma and related bone disease. Ann Transl Med. 2015;3(21):334.
Sato S, Katsushima K, Shinjo K, Hatanaka A, Ohka F, Suzuki S, Naiki-Ito A, Soga N, Takahashi S, Kondo Y. Histone deacetylase inhibition in prostate Cancer triggers miR-320-mediated suppression of the androgen receptor. Cancer Res. 2016;76(14):4192–204.
Shuliang S, Lei C, Guangwu J, Changjie L. Involvement of ubiquitin-conjugating enzyme E2C in proliferation and invasion of prostate carcinoma cells. Oncol Res. 2013;21(3):121–7.
Chen Z, Zhang C, Wu D, Chen H, Rorick A, Zhang X, Wang Q. Phospho-MED1-enhanced UBE2C locus looping drives castration-resistant prostate cancer growth. EMBO J. 2011;30(12):2405–19.
Hsieh IS, Chang KC, Tsai YT, Ke JY, Lu PJ, Lee KH, Yeh SD, Hong TM, Chen YL. MicroRNA-320 suppresses the stem cell-like characteristics of prostate cancer cells by downregulating the Wnt/beta-catenin signaling pathway. Carcinogenesis. 2013;34(3):530–8.
Leung CM, Li SC, Chen TW, Ho MR, Hu LY, Liu WS, Wu TT, Hsu PC, Chang HT, Tsai KW. Comprehensive microRNA profiling of prostate cancer cells after ionizing radiation treatment. Oncol Rep. 2014;31(3):1067–78.
Kong L, Schafer G, Bu H, Zhang Y, Zhang Y, Klocker H. Lamin a/C protein is overexpressed in tissue-invading prostate cancer and promotes prostate cancer cell growth, migration and invasion through the PI3K/AKT/PTEN pathway. Carcinogenesis. 2012;33(4):751–9.
Acknowledgements
We thank Melissa Stauffer, PhD, for editing the manuscript.
Funding
This work has been supported in part by the Center of Genomic and Precision medicine, National Taiwan University, Taiwan, with the grant number 106R8400, and the Center for Biotechnology, National Taiwan University, Taiwan, with the grant number GTZ300. The funders had no role in the design of the study; in the collection, analysis or interpretation of data; in writing the manuscript; or in the decision to submit the manuscript for publication.
Availability of data and materials
The source code and manual of the anamiR package are available at https://bioconductor.org/packages/release/bioc/html/anamiR.html.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: TPL and EYC. Wrote the anamiR package and analyzed the data: TTW. Constructed the database: TTW and CYL. Contributed materials and analysis tools: LCL, MHT, and EYC. Wrote the paper: TTW, TPL, and CYL. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1
Table S1. The total potential number of miRNA-gene pairs obtained by tallying different prediction algorithms. Table S2. Number of miRNA/gene interaction pairs in the prediction algorithms and experimentally validated databases included in the anamiR package. Table S3. Characteristics of anamiR and miRComb. Table S4. Pairs with negative correlation coefficients (GSE16558). Table S5. Pairs with negative correlation coefficients (GSE60371). Table S6. The default parameters used in the examples. Table S7. Top 5 interaction pairs with negative correlation coefficients in the 5 pathways identified by function-driven analysis. (DOCX 38 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, TT., Lee, CY., Lai, LC. et al. anamiR: integrated analysis of MicroRNA and gene expression profiling. BMC Bioinformatics 20, 239 (2019). https://doi.org/10.1186/s12859-019-2870-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-019-2870-x