
Abstract
Background
Host–pathogen interactions are important in a wide range of research fields. Given the importance of metabolic crosstalk between hosts and pathogens, a metabolic network-based reverse ecology method was proposed to infer these interactions. However, the validity of this method remains unclear because of the various explanations presented and the influence of potentially confounding factors that have thus far been neglected.
Results
We re-evaluated the importance of the reverse ecology method for evaluating host–pathogen interactions while statistically controlling for confounding effects using oxygen requirement, genome, metabolic network, and phylogeny data. Our data analyses showed that host–pathogen interactions were more strongly influenced by genome size, primary network parameters (e.g., number of edges), oxygen requirement, and phylogeny than the reserve ecology-based measures.
Conclusion
These results indicate the limitations of the reverse ecology method; however, they do not discount the importance of adopting reverse ecology approaches altogether. Rather, we highlight the need for developing more suitable methods for inferring host–pathogen interactions and conducting more careful examinations of the relationships between metabolic networks and host–pathogen interactions.
Similar content being viewed by others

Background
Diseases spread in natural host (e.g., human and plant) populations via pathogens. Investigations of host–pathogen interactions are important not only in the context of basic scientific research but also in applied biological research fields such as medical science and disease ecology [1,2,3]. The development and progress of several new technologies and high-throughput methods have generated considerable host–pathogen interaction data, which have accumulated in several databases such as the Pathogen-Host Interactions database (PHI-base) [4] and Host Pathogen Interaction Database [5].
Elucidating the molecular mechanisms of host–pathogen interactions is important for host–pathogen interaction inference; in particular, pathogens use their biomolecules to hijack and re-wire numerous biochemical pathways in their hosts during infection [6]. Recognition of the importance of metabolic crosstalk between hosts and pathogens led to the proposal of a reverse ecology approach based on metabolic networks [7] as a computational framework for estimating host–pathogen interactions, which has attracted increasing attention [8]. Metabolism, a series of chemical reactions, is often represented as a network (known as a metabolic network). Metabolic networks have mainly been studied from a complex network perspective given the advances in network science [9, 10], especially network biology [11]. Indeed, many studies have evaluated adaptations to different environments (i.e., ecological interactions) by examining metabolic networks [12,13,14]. Specifically, Lévy et al. [15] used a graph theoretical algorithm to identify the set of exogenously acquired nutrients (known as a seed set) in metabolic networks, and proposed measures for estimating the cooperative interactions between a species pair [16, 17]: the biosynthetic support score (BSS) and the metabolic complementarity index (MCI). The BSS quantifies the metabolic ability of an organism (e.g., host) to meet the nutritional requirements of another organism (e.g., pathogen) [16]. The MCI indicates the degree of support one organism provides to another organism through biosynthetic complementarity (i.e., potential for syntrophism). Although the authors [16] stated that the MCI is particularly useful for estimating pairwise interactions between co-occurring microbes, it is also expected to be useful for assessing host–pathogen interactions because of the common occurrence of pathogenic symbiosis in plants [18] and insects [19]. A previous study [17] showed that these measures (particularly the BSS) were effective for predicting host–pathogen interactions. The reverse ecology method has been implemented as a software [16] and R-package [20], and has been applied in several microbial ecology studies such as studies of the human gut microbiome (e.g., [21, 22]).
However, more careful examination may be required to determine the importance of reverse ecology-based measures (i.e., BSS and MCI) on host–pathogen interaction inference. In particular, previous studies did not take several alternative factors into account. For example, genome size and total gene number were not directly evaluated, although it is well-known that these genomic parameters of pathogens are lower than those of free-living microbes [23]. The oxygen requirement of pathogens has also been omitted in previous models, despite the importance of oxygen in host–pathogen interactions [24] (i.e., pathogens exhibit remarkable adaptability and prevail in a wide range of oxygen concentrations); in addition, metabolic networks of aerobes are larger and less modular (or compartmentalized) than those of anaerobes [25, 26]. The effect of metabolic network modularity on host–pathogen interactions has not yet been evaluated, although previous studies [27, 28] showed that the metabolic network modularity of obligate host-associated bacteria was lower than that of free-living bacteria. In turn, genomic, physiological, and network parameters may influence the BSS and MCI values; thus, controlling for these potentially confounding effects is necessary to determine the importance and relevance of the BSS and MCI. However, previous studies did not control for these confounding effects. More importantly, the effects of phylogenetic signals were not considered, although the importance of phylogeny in evaluating associations between biological features has been well-established through comparative phylogenetic analyses [29, 30]. For example, an opposite conclusion may be derived when considering comparative phylogenetic analysis [31, 32].
Thus, we re-evaluated the contribution of the parameters BSS and MCI to pathogen/non-pathogen classification while statistically controlling for potentially confounding effects using data related to oxygen requirement, genome, and metabolic networks. We also performed comparative phylogenetic analyses to evaluate the effects of phylogenetic signals on the association between reverse ecology-based measures and host–pathogen interactions.
Methods
Host–pathogen interactions
Host–pathogen interaction data were downloaded from PHI-base (www.phi-base.org) [4] on July 28, 2016. Pathogenic species were chosen based on the availability of metabolic network data in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database [33] and information related to oxygen requirement in the Microbial Physiology and Metabolism (MIPMET) database (takemoto08.bio.kyutech.ac.jp/mipmet/); 54 mammalian pathogens, 13 plant pathogens, and 15 insect pathogens were selected (Additional file 1). The classification of mammalian/plant/insect pathogens was defined based on the information of Host Description (i.e., host classification) for each pathogen in the XML file downloadable from PHI-base. Specifically, the host species of mammalian pathogens are categorized into Rodents, Rabbits & Hares, Primates, Odd-toed Ungulates, and Even-toed Ungulates. The host species of plant pathogens are classified into Eudicots, Flowering Plants, and Monocots. Host species insects are classified as Bees, Beetles, Flies, Black-legged Ticks, Moths, and Fleas.
Non-pathogenic species
We defined 273 candidate non-pathogenic species based on microbial physiology and metabolism data (i.e., lifestyle, habitat, and growth temperature) (Additional file 2). Data related to microbial physiology and metabolism were collected from the literature (e.g., [25, 26, 34]) and are available in the MIPMET database. The datasets for microbial physiology and metabolism were downloaded from the database on August 25, 2016. We first selected species that were classified both as Free-living in the Biotic category and as Mesophilic in the Temperature category, while species classified as Host-associated in the Habitat category were ignored. We next removed species whose genera appeared in the PHI-base dataset. Finally, we only selected species whose oxygen requirement data were available in the database.
Biosynthetic support score and metabolic complementarity index
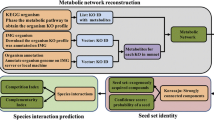
The BSS and MCI values between species were calculated using NetCooperate software [16], downloaded from the website (depts.washington.edu/elbogs/NetCooperate/NetCooperateWeb.cgi) on September 2, 2016. The BSS is defined as the fraction of the seed set of an organism that is available in the metabolic network of another organism. The MCI is defined as the fraction of the seed set of an organism that is available in the non-seed set of another organism. Both the BSS and MCI range from 0 (no potential for cooperation) to 1 (perfect cooperation). The metabolic networks, required for the software, were constructed according to previous studies [17, 25]. XML files (version 0.7.1) containing metabolic network data (i.e., substrate–product relationships and reversibility/irreversibility of chemical reactions) were downloaded from the KEGG database [33] (ftp://ftp.genome.jp/pub/kegg/xml/kgml/metabolic/organisms/) on August 26, 2016. Based on the XML files, metabolic networks were represented as directed networks, in which the nodes and edges correspond to metabolites and reactions (i.e., substrate–product relationships), respectively. Because the use of such data may be desirable to ensure reproducibility, the present dataset on metabolic networks is available upon request. When calculating the BSS and MCI between hosts and microbes, we focused on representative host species whose metabolic pathways have been well-characterized using experimental approaches, because the metabolic networks of hosts registered in PHI-base may be not available in the KEGG database; specifically, we used the metabolic networks of Homo sapiens (human), Arabidopsis thaliana (thale cress), and Drosophila melanogaster (fruit fly) for mammal, plant, and insect host species, respectively. The BSS and MCI are asymmetric between a species pair [16] (i.e., host and microbe, in this study); thus, we considered two types of BSS and MCI values, respectively: we calculated scores for the biosynthetic support of a microbe for a host (BSSMH), biosynthetic support of a host for a microbe (BSSHM), biosynthetic complement of a microbe for a host (MCIMH), and biosynthetic complement of a host for a microbe (MCIHM).
Genomic and network parameters
For microbes, we obtained the genome size and number of total protein-encoding genes from the KEGG database on October 30, 2016. As network parameters, we evaluated the number of nodes (N) and number of directed edges (E). We focused on network modularity, since a previous study [28] demonstrated its importance on pathogen/non-pathogen classification. The modularity of networks is often measured using the Q-value (e.g., [35]). Q is defined as the fraction of edges that lie within, rather than between, modules relative to that expected by chance. The Q-value is a size-invariant measure; thus, the role of network size on modularity can be analyzed as an independent topological variable of interest [28] (however, see [36]). A network with a higher Q-value indicates a higher modular structure. Thus, we need to find the global maximum Q-value over all possible divisions. Since it is hard to find the optimal division with the maximum Q in general, approximate optimization techniques are required. In this study, a spectral optimization method was used for directed networks [37, 38] to avoid the resolution limit problem in community (or module) detection [35, 39] as much as possible.
Statistical analysis
To evaluate the contribution of each parameter (or factor) to pathogen/non-pathogen classification, we conducted logistic regression analyses using R software (version 3.3.2; www.R-project.org). There was no biological replicate in our dataset (see also Additional file 1). The ordinary logistic regression based on fixed effects was first considered, for which we constructed full models encompassing the given explanatory variables, and selected the best model based on the sample size-corrected version of Akaike information criterion (AICc) values using the R package MuMIn (version 1.15.6). The quantitative variables were normalized to the same scale, with a mean of 0 and standard deviation of 1, using the scale function in R before the analysis. We used the power.roc.test function in the R package pROC (version 1.9.1) to estimate the required sample size based on the area under the receiver operating characteristic curve (AUC) value of the best model, statistical power, and balance between control and case observations (i.e., non-pathogens and pathogens). To avoid model selection bias, we also adopted a model-averaging approach [40], from which we obtained the averaged models in the top 95% confidence set of models using the model.avg function in the R package MuMIn. Genome size and total gene number were log-transformed for all analyses.
To remove the effects of phylogenetic signals from the regression analyses, we performed phylogenetic logistic regression analyses using the function phyloglm in the R-package phylolm (version 2.5). The phylogenetic trees, which are required for phylogenetic regression, were constructed using 16S rRNA sequence data according to the all-species living tree project [41] (Additional files 3, 4 and 5). 16S rRNA gene sequences were obtained from the KEGG database on November 30, 2016. After multiple alignments of the nucleotide sequences using ClustalW2 software, the phylogenetic tree was constructed using NJplot (doua.prabi.fr/software/njplot). Similar to our approach for logistic regression analyses, we constructed full models and then selected the best model based on AICc values. We also obtained the averaged models.
The contribution (i.e., non-zero estimate) of each explanatory variable to the pathogen/non-pathogen dichotomy was considered to be complete when the associated p-value was less than 0.05.
Results and Discussion
Re-evaluation of the metabolic network-based reverse ecology method
The conditions for the present data analysis may differ from those used in the previous study [17]. For example, the pathogen and non-pathogen datasets may differ between this study and the previous study because the dataset was not clearly described in the previous study. Metabolic networks may also differ between this study and the previous study because the database has been updated. To determine whether the differences in analytical conditions were not limiting, we first evaluated the validity of the reverse ecology method under similar conditions as those used in the previous study; that is, we performed statistical analysis using only the BSS (BSSHM and BSSMH) and MCI (MCIHM and MCIMH) values. We then determined the contributions of the BSSs and MCIs to pathogen/non-pathogen classification (Table 1). Our results were similar to those of the previous study and were consistent with empirical evidence. In particular, biosynthetic support of hosts for microbes (BSSHM) was observed in host–pathogen interactions; however, biosynthetic support of microbes for hosts (BSSMH) was negatively or not associated with the interactions. This result reflects the parasitism of pathogens (i.e., pathogens benefit from hosts, while hosts do not benefit from pathogens). For plants and insects, the biosynthetic complement of microbes for hosts (MCIMH) was observed in the host–pathogen interactions because of pathogenic symbiosis in plants [18] and insects [19]. The biosynthetic complement of the hosts for microbes (MCIHM) showed a certain degree of negative contribution to the pathogen/non-pathogen classification. This indicates that pathogens avoid benefiting from hosts in the context of biosynthetic complementation. This result is puzzling; however, it may be explained as follows. MCIHM is defined as the fraction of the seed set of a microbe that is available in the non-seed set of a host, whereas BSSHM is the fraction of the seed set of the microbe available in all metabolites (i.e., union of the seed set and non-seed set) of the host. Thus, the negative effect of MCIHM despite the positive effect of BSSHM indicates that the seed set of the microbe is mainly supported by the seed set of the host. This suggests competition between hosts and microbes (i.e., microbes consume the nutrients required by the host), which is a parasitic property.
Effects of genomic, physiological, and network parameters
We aimed to confirm the contributions of the BSS and MCI to pathogen/non-pathogen classification. However, the validity of the BSS and MCI remains controversial; this is because of other factors that may dominantly contribute to pathogen/non-pathogen classification, as described in the Background section. Thus, we next constructed full models encompassing all explanatory variables (BSSHM, BSSMH, MCIHM, MCIMH, genome size, total gene number, oxygen requirement, N, E, and Q) to control for potentially confounding effects. The AICc values in the best models generally decreased because of the consideration of the physiological, genomic, and primary network parameters (Tables 1 and 2). This indicates the importance of consideration of these parameters. The averaged models showed that host–pathogen interactions were affected by the oxygen requirement (i.e., anaerobic or not) and primary network parameters (i.e., N and E) of microbial metabolic networks rather than by the BSS and MCI, although these metabolic network-based reverse ecology parameters were found to partly contribute to the best models (Table 2). This is partly because the BSS and MCI are strongly related to the other parameters. In mammalian pathogens, for example, BSSHM is positively correlated with N (Spearman’s rank correlation coefficient r s = 0.94, p < 2.2 × 10−16) and E (r s = 0.94, p < 2.2 × 10−16). MCIHM is also positively associated with with N (r s = 0.84, p < 2.2 × 10−16) and E (r s = 0.84, p < 2.2 × 10−16). Empirical evidence supports these results. In particular, mammalian pathogens are generally facultative or strictly aerobes. This is consistent with the observation that pathogens must adapt to varied oxygen concentrations [24]. Mammalian pathogens show smaller genome sizes, and both mammalian and insect pathogens have relatively smaller metabolic networks. This indicates the minimalism of pathogens [23]. However, the previous study [17] showed that the number of nodes had a limited effect on pathogen/non-pathogen prediction using receiver operating characteristic curves. This discrepancy between the present and previous study is related to the use of different analysis methods. The receiver operating characteristic-based analyses used in the previous study did not control for confounding effects; thus, the effect of the number of nodes was likely underestimated. Moreover, the previous study did not evaluate the effect of another primary network parameter: the number of edges. Pathogens have a relatively large number of directed edges, indicating that the metabolic networks of the pathogens are relatively dense. This may be because many metabolic pathways, except for central metabolism (such as energy metabolism), in pathogens depend on host species metabolism [42, 43]. Pathogens lack peripheral metabolic pathways (e.g., lipid metabolism and amino acid metabolism), which is supported by the importance of amino acids on host–pathogen metabolic interactions [8] and is consistent with the conclusion of a bioinformatics study on the pathway-based inference of host–pathogen interactions [44]. Metabolic networks exhibit a bow-tie (or core–peripheral) structure [45]: they can be decomposed into densely connected giant components (core) and thinly connected peripheral subnetworks. Central metabolism is located at the core; thus, metabolic networks of pathogens are denser than those of non-pathogens because they only consist of densely connected components. In contrast to the previous studies [27, 28], metabolic network modularity did not differ between pathogens (or host-associated species) and non-pathogens, which is in line with the conclusion of other studies. In particular, the size of the metabolic network is a major determinant of network modularity [46]; that is, the difference in metabolic networks between pathogens and non-pathogens is explained by network size (i.e., N and E) rather than network modularity. Furthermore, the previously observed difference in network modularity between host-associated species and free-living species was probably due to a lack of available data on metabolic reactions; rather, metabolic network modularity was found to be dependent on species growth conditions such as oxygen requirement [47]. These previous studies also support the importance of the oxygen requirement and primary network parameters. However, it remains possible that the observed limited effect of the BSS and MCI is due to the sample size; in particular, our dataset contained only 13 plant pathogens and 15 insect pathogens; thus, statistical power for detecting an effect may be low. However, the AUC values obtained from the best models in the cases of plant pathogens and insect pathogens were relatively high at 0.844 and 0.905, respectively. When the statistical power of 0.95 was considered, the required sample sizes of plant pathogens and insect pathogens were 9 and 6, respectively. This result indicates that the sample sizes pose few problems.
Effect of phylogenetic signals
As described in the Background, it is important to consider the effects of phylogenetic signals when investigating the associations between biological features. We removed the phylogenetic effects using phylogenetic logistic regression. The AICc values in the best models generally decreased with consideration of the phylogeny (Tables 2 and 3), indicating the importance of phylogeny. Again, the averaged models revealed the limited effects of the BSS and MCI on pathogen/non-pathogen classification (Table 3). Moreover, the averaged models showed that the other parameters were only minimally associated with host–pathogen interactions; however, clear contributions of primary network parameters (i.e., N and E) were observed in the case of insect pathogens. According to the best models, each parameter partly contributes to pathogen/non-pathogen classification. For example, the genome sizes of mammalian pathogens were smaller than those of non-pathogens, and the metabolic networks of mammalian pathogens were denser than those of non-pathogens. In addition, biosynthetic complementation of the microbes for the insect host was observed. Insect pathogens are typically aerobic. However, the averaged models showed that these results were not statistically robust. The difference between the best model and averaged model was due to model selection bias. These results indicate phylogenetic bias in host–pathogen interactions (i.e., phylogenetic information, rather than reverse ecology-based measures and other parameters, determines whether a species is pathogenic). The effect of phylogenetic signals (the fact that important biological associations were not conclusively determined with phylogenetic correction) has been observed in a wide range of research fields (e.g., in metabolic networks [31] and in species–species interactions in food webs [32]). However, more careful examinations are required because of the limitations of phylogenetic comparative analysis. In particular, phylogenetic comparative analysis assumes a Brownian motion-like evolution of biological traits on a phylogenetic tree with accurate branch lengths, and thus may result in misleading conclusions. We constructed the phylogenetic trees based on 16S rRNA sequences only to reduce computational costs. Ideally, a highly resolved phylogenic tree [48] constructed based on a common protein set across organisms may be required. In addition, statistical power decreases when a dataset is reduced in size following phylogenetic corrections [49]. As mentioned in the previous section, our dataset contained only a few samples for plant pathogens and insect pathogens; thus, statistical power may have been low. Ideally, the sample sizes required for suitable statistical power would be evaluated. However, methods for estimating the sample sizes have not yet been established for the phylogenetic logistic regression model. Thus, more careful examinations are required to determine the limited effect of the BSS and MCI. In this context, a larger dataset of host–pathogen interactions should be evaluated. The development of high-throughput sequencing techniques will enable the collection of such data. For example, metagenomic techniques can now reveal host–pathogen interactions [50]. Similar to numerous previous studies of host–pathogen interactions, our study was limited because of the lack of availability of accurate datasets for non-pathogenic species (i.e., negative set) owing to the lack of experimental evidence, although we avoided this limitation as much as possible by using data related to microbial physiology and metabolism. Metagenomic techniques may also enable acquisition of a more accurate dataset.
Conclusions
The results presented herein call into question the importance of the current version of the metabolic network-based reverse ecology approach (i.e., BSS and MCI) for host–pathogen interaction inference. Metabolic networks are still not fully understood in detail. For example, enzyme promiscuity [51], which implies that enzymes can catalyze multiple reactions, act on more than one substrate, or exert a range of suppressions (in which enzymatic function is suppressed by over-expressing enzymes with originally different functions [52]), suggests the existence of many hidden metabolic reactions. Consideration of these hidden metabolic reactions is important for understanding metabolic interactions in ecosystems. However, the results of the present study do not entirely discount the metabolic network-based reverse ecology approach; rather, these findings emphasize the need for developing more suitable methods for estimating host–pathogen interactions. For example, the definition of seed sets is controversial. Previous studies [15, 17] used a strongly connected component decomposition algorithm to identify a seed set. However, this method only focuses on network topology and does not consider biochemically feasible reactions. For example, it may be necessary to identify seed sets based on an algorithm of network expansion to generate the set of all possible metabolites that can be produced from a set of compounds, similar to the approach adopted in a previous study [53]. An approach for pathway-based inference of host–pathogen interactions [44] would also be useful, which would allow for more careful comparisons of metabolic networks between hosts and pathogens. Moreover, there are several metabolic models based on flux balance analysis. Originally, flux balance analysis was used to model metabolic processes in single species; however, in recent years, this method has started to be applied in microbial ecology (e.g., to examine the cooperative and competitive dynamics between different species) [54,55,56,57,58]. These methods can improve understanding of interspecies interactions at the metabolic level, although the computational costs are higher compared to those required with the reverse ecology method. Metabolic network-based reverse ecology remains a challenging research topic in the post-genomic era because of the importance of the human microbiome [59] and the earth microbiome [60]; thus, more careful investigations of the relationships between metabolic networks and host–pathogen interactions are needed.
Abbreviations
- AICc:
-
Sample size-corrected version of Akaike Information Criterion
- AUC:
-
Area Under the receiver operating characteristic Curve
- BSS:
-
Biosynthetic support score
- BSSHM :
-
Biosynthetic support score of a host for a microbe
- BSSMH :
-
Biosynthetic support score of a microbe for a host
- E:
-
number of edges
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- MCI:
-
Metabolic complementarity index
- MCIHM :
-
Biosynthetic complement index of a host for a microbe
- MCIMH :
-
Biosynthetic complement index of a microbe for a host
- MIPMET:
-
MIcrobial Physiology and METabolism
- N:
-
Number of nodes
- PHI-base:
-
Pathogen-host interactions database
References
Engering A, Hogerwerf L, Slingenbergh J. Pathogen-host-environment interplay and disease emergence. Emerg Microbes Infect. 2013;2:e5. Available from: http://dx.doi.org/10.1038/emi.2013.5.
Pilosof S, Fortuna MA, Cosson J-F, Galan M, Kittipong C, Ribas A, et al. Host-parasite network structure is associated with community-level immunogenetic diversity. Nat Commun. 2014;5:5172. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25312328.
Johnson PTJ, Preston DL, Hoverman JT, Lafonte BE. Host and parasite diversity jointly control disease risk in complex communities. Proc Natl Acad Sci U S A. 2013;110:16916–21. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24082092.
Urban M, Pant R, Raghunath A, Irvine AG, Pedro H, Hammond-Kosack KE. The Pathogen-Host Interactions database (PHI-base): additions and future developments. Nucleic Acids Res. 2015;43:D645–55. Available from: http://nar.oxfordjournals.org/lookup/doi/10.1093/nar/gku1165.
Ammari MG, Gresham CR, McCarthy FM, Nanduri B. HPIDB 2.0: a curated database for host–pathogen interactions. Database. 2016;2016. Available from: http://database.oxfordjournals.org/lookup/doi/10.1093/database/baw103.
Shah PS, Wojcechowskyj JA, Eckhardt M, Krogan NJ. Comparative mapping of host–pathogen protein–protein interactions. Curr Opin Microbiol. 2015;27:62–8. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1369527415000995.
Levy R, Borenstein E. Reverse ecology: from systems to environments and back. Adv Exp Med Biol. 2012;751:329–45. [cited 2012 Sep 26]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/22821465.
Olive AJ, Sassetti CM. Metabolic crosstalk between host and pathogen: sensing, adapting and competing. Nat Rev Microbiol. 2016;14:221–34. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26949049.
Barabási A-L. Network science. Philos Trans R Soc A. 2013;371:20120375. Available from: http://rsta.royalsocietypublishing.org/content/371/1987/20120375.
Takemoto K, Oosawa C. Introduction to complex networks: measures, statistical properties, and models. Stat. Mach. Learn. Approaches Netw. Anal. 2012. p. 45–75.
Barabási A-L, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5:101–13. [cited 2012 Apr 8]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/14735121.
Takemoto K. Current understanding of the formation and adaptation of metabolic systems based on network theory. Metabolites. 2012;2:429–57.
Papp B, Teusink B, Notebaart RA. A critical view of metabolic network adaptations. HFSP J. 2009;3:24–35. [cited 2011 Jun 14]. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2689614/.
Nam H, Conrad TM, Lewis NE. The role of cellular objectives and selective pressures in metabolic pathway evolution. Curr Opin Biotechnol. 2011;22:595–600. [cited 2012 Mar 15]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/21481583.
Borenstein E, Kupiec M, Feldman MW, Ruppin E. Large-scale reconstruction and phylogenetic analysis of metabolic environments. Proc Natl Acad Sci U S A. 2008;105:14482–7. Available from: http://www.pnas.org/content/105/38/14482.
Levy R, Carr R, Kreimer A, Freilich S, Borenstein E. NetCooperate: a network-based tool for inferring host-microbe and microbe-microbe cooperation. BMC Bioinformatics. 2015;16:164. Available from: http://www.biomedcentral.com/1471-2105/16/164.
Borenstein E, Feldman MW. Topological signatures of species interactions in metabolic networks. J Comput Biol. 2009;16:191–200. [cited 2012 Mar 19] Available from: http://www.ncbi.nlm.nih.gov/pubmed/19178139.
de Souza EM, Granada CE, Sperotto RA. Plant pathogens affecting the establishment of plant-symbiont interaction. Front Plant Sci. 2016;7:15. Available from: http://journal.frontiersin.org/Article/10.3389/fpls.2016.00015/abstract.
Abebe E, Abebe-Akele F, Morrison J, Cooper V, Thomas WK. An insect pathogenic symbiosis between a Caenorhabditis and Serratia. Virulence. 2011;2:158–61. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3100764/.
Cao Y, Wang Y, Zheng X, Li F, Bo X. RevEcoR: an R package for the reverse ecology analysis of microbiomes. BMC Bioinformatics. 2016;17:294. Available from: http://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-016-1088-4.
Freilich S, Zarecki R, Eilam O, Segal ES, Henry CS, Kupiec M, et al. Competitive and cooperative metabolic interactions in bacterial communities. Nat Commun. 2011;2:589. [cited 2012 Mar 12]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/22158444.
Zelezniak A, Andrejev S, Ponomarova O, Mende DR, Bork P, Patil KR. Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proc Natl Acad Sci. 2015;112:6449–54. Available from: http://www.pnas.org/content/112/20/6449.
Moran NA. Microbial minimalism. Cell. 2002;108:583–6. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0092867402006657.
Marteyn B, Scorza FB, Sansonetti PJ, Tang C. Breathing life into pathogens: the influence of oxygen on bacterial virulence and host responses in the gastrointestinal tract. Cell Microbiol. 2011;13:171–6. Available from: http://doi.wiley.com/10.1111/j.1462-5822.2010.01549.x.
Takemoto K, Borjigin S. Metabolic network modularity in Archaea depends on growth conditions. PLoS One. 2011;6:e25874. [cited 2012 Apr 6]; Available from: http://dx.plos.org/10.1371/journal.pone.0025874.
Mazurie A, Bonchev D, Schwikowski B, Buck GA. Evolution of metabolic network organization. BMC Syst Biol. 2010;4:59. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20459825.
Parter M, Kashtan N, Alon U. Environmental variability and modularity of bacterial metabolic networks. BMC Evol Biol. 2007;7:169. [cited 2011 Jun 11]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/17888177.
Kreimer A, Borenstein E, Gophna U, Ruppin E. The evolution of modularity in bacterial metabolic networks. Proc Natl Acad Sci U S A. 2008;105:6976–81. Available from: http://www.pnas.org/content/105/19/6976.
Garland T, Bennett AF, Rezende EL. Phylogenetic approaches in comparative physiology. J Exp Biol. 2005;208:3015–35. [cited 2013 May 21]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/16081601.
Garland T, Harvey PH, Ives AR. Procedures for the analysis of comparative data using phylogenetically independent contrasts. Syst Biol. 1992;41:18–32. [cited 2013 May 29] Available from: http://sysbio.oxfordjournals.org/cgi/doi/10.1093/sysbio/41.1.18.
Takemoto K, Yoshitake I. Limited influence of oxygen on the evolution of chemical diversity in metabolic networks. Metabolites. 2013;3:979–92. [cited 2013 Oct 17]; Available from: http://www.mdpi.com/2218-1989/3/4/979/.
Naisbit RE, Kehrli P, Rohr RP, Bersier L-F. Phylogenetic signal in predator–prey body-size relationships. Ecology. 2011;92:2183–9. Available from: http://doi.wiley.com/10.1890/10-2234.1.
Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–62. Available from: http://nar.oxfordjournals.org/lookup/doi/10.1093/nar/gkv1070.
Freilich S, Kreimer A, Meilijson I, Gophna U, Sharan R, Ruppin E. The large-scale organization of the bacterial network of ecological co-occurrence interactions. Nucleic Acids Res. 2010;38:3857–68. [cited 2012 Mar 9] Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2896517/.
Fortunato S. Community detection in graphs. Phys Rep. 2010;486:75–174. [cited 2012 Mar 9] Available from: http://linkinghub.elsevier.com/retrieve/pii/S0370157309002841.
Guimerà R, Sales-Pardo M, Amaral L. Modularity from fluctuations in random graphs and complex networks. Phys Rev E. 2004;70:025101(R). [cited 2011 Jul 22]; Available from: http://link.aps.org/doi/10.1103/PhysRevE.70.025101.
Leicht E, Newman M. Community structure in directed networks. Phys Rev Lett. 2008;100:118703. [cited 2013 May 23]; Available from: http://link.aps.org/doi/10.1103/PhysRevLett.100.118703.
Clune J, Mouret J-B, Lipson H. The evolutionary origins of modularity. Proc R Soc B. 2013;280:20122863. [cited 2013 Mar 1] Available from: http://www.ncbi.nlm.nih.gov/pubmed/23363632.
Fortunato S, Barthélemy M. Resolution limit in community detection. Proc Natl Acad Sci U S A. 2007;104:36–41. Available from: http://www.pnas.org/content/104/1/36.
Takemoto K, Kajihara K. Human impacts and climate change influence nestedness and modularity in food-web and mutualistic networks. PLoS One. 2016;11:e0157929. Available from: http://dx.plos.org/10.1371/journal.pone.0157929.
Yarza P, Richter M, Peplies J, Euzeby J, Amann R, Schleifer K-H, et al. The All-Species Living Tree project: a 16S rRNA-based phylogenetic tree of all sequenced type strains. Syst Appl Microbiol. 2008;31:241–50. [cited 2011 Jul 22]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/18692976.
Takemoto K, Niwa T, Taguchi H. Difference in the distribution pattern of substrate enzymes in the metabolic network of Escherichia coli, according to chaperonin requirement. BMC Syst Biol. 2011;5:98. [cited 2011 Jul 29] Available from: http://www.ncbi.nlm.nih.gov/pubmed/21702926.
Glass JI, Lefkowitz EJ, Glass JS, Heiner CR, Chen EY, Cassell GH. The complete sequence of the mucosal pathogen Ureaplasma urealyticum. Nature. 2000;407:757–62. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11048724.
Kleftogiannis D, Wong L, Archer JAC, Kalnis P. Hi-Jack: a novel computational framework for pathway-based inference of host–pathogen interactions. Bioinformatics. 2015;31:2332–9. Available from: http://bioinformatics.oxfordjournals.org/lookup/doi/10.1093/bioinformatics/btv138.
Ma H-W, Zhao X-M, Yuan Y-J, Zeng A-P. Decomposition of metabolic network into functional modules based on the global connectivity structure of reaction graph. Bioinformatics. 2004;20:1870–6. [cited 2013 Feb 28]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/15037506.
Zhou W, Nakhleh L. Convergent evolution of modularity in metabolic networks through different community structures. BMC Evol Biol. 2012;12:181. Available from: http://bmcevolbiol.biomedcentral.com/articles/10.1186/1471-2148-12-181.
Takemoto K. Does habitat variability really promote metabolic network modularity? PLoS One. 2013;8:e61348. [cited 2013 Apr 15]; Available from: http://dx.plos.org/10.1371/journal.pone.0061348.
Ciccarelli FD, Doerks T, von Mering C, Creevey CJ, Snel B, Bork P. Toward automatic reconstruction of a highly resolved tree of life. Science. 2006;311:1283–7. [cited 2013 Jun 24]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/16513982.
Griffith OL, Moodie GEE, Civetta A. Genome size and longevity in fish. Exp Gerontol. 2003;38:333–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12581799.
Koch L. Metagenomics: pathogen diagnostics for the masses. Nat Rev Genet. 2016;17:378. Available from: http://www.nature.com/doifinder/10.1038/nrg.2016.76.
Khersonsky O, Roodveldt C, Tawfik DS. Enzyme promiscuity: evolutionary and mechanistic aspects. Curr Opin Chem Biol. 2006;10:498–508. [cited 2011 Jul 7]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/16939713.
Patrick WM, Quandt EM, Swartzlander DB, Matsumura I. Multicopy suppression underpins metabolic evolvability. Mol Biol Evol. 2007;24:2716–22. [cited 2011 Jul 22]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/17884825.
Handorf T, Christian N, Ebenhöh O, Kahn D. An environmental perspective on metabolism. J Theor Biol. 2008;252:530–7. [cited 2013 Aug 6]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/18086477.
Lewis NE, Nagarajan H, Palsson BO. Constraining the metabolic genotype-phenotype relationship using a phylogeny of in silico methods. Nat Rev Microbiol. 2012;10:291–305. [cited 2012 May 24]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/22367118.
Klitgord N, Segrè D. Ecosystems biology of microbial metabolism. Curr Opin Biotechnol. 2011;22:541–6. [cited 2012 Mar 2]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/21592777.
Harcombe WR, Riehl WJ, Dukovski I, Granger BR, Betts A, Lang AH, et al. Metabolic resource allocation in individual microbes determines ecosystem interactions and spatial dynamics. Cell Rep. 2014. [cited 2014 May 24]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/24794435.
Klitgord N, Segrè D. Environments that induce synthetic microbial ecosystems. PLoS Comput Biol. 2010;6:e1001002. [cited 2012 May 15]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/21124952.
Magnúsdóttir S, Heinken A, Kutt L, Ravcheev DA, Bauer E, Noronha A, et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat Biotechnol. 2016. Available from: http://dx.doi.org/10.1038/nbt.3703.
Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet. 2012;13:260–70. Available from: http://www.nature.com/doifinder/10.1038/nrg3182.
Gilbert JA, Jansson JK, Knight R. The earth microbiome project: successes and aspirations. BMC Biol. 2014;12:69. Available from: http://bmcbiol.biomedcentral.com/articles/10.1186/s12915-014-0069-1.
Acknowledgments
The authors thank J.-B. Mouret for providing an executable file for calculating Q.
Funding
This study was supported by JSPS KAKENHI Grant Numbers JP25700030 and JP17H04703. The funding body had no role in the design, collection, analysis or interpretation of this study.
Availability of data and materials
All data analyzed during this study are included in this published article and its supplementary information files.
Authors’ contributions
KT conceived and designed the study. KT and KA prepared the data and performed data analysis. Both authors interpreted the results. KT drafted the manuscript. Both authors gave final approval for publication.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
List of pathogens used in this study. This table shows the species name, Kyoto Encyclopedia of Genes and Genomes (KEGG) organism identifier (see www.genome.jp/kegg/catalog/org_list.html), host classification, oxygen requirement, genome size [bp], number of total genes [count], BSSHM, BSSMH, MCIHM, MCIMH, number of nodes, number of directed edges, and network modularity Q (XLSX 45 kb).
Additional file 2:
List of non-pathogens used in this study. See the Additional file 1 caption for a description of table elements (XLSX 65 kb).
Additional file 3:
Phylogenetic tree of mammalian pathogens and non-pathogens used in this study. Node labels correspond to the KEGG organism identifier. The tree is presented in the Newick format (TXT 6 kb).
Additional file 4:
Phylogenetic tree of plant pathogens and non-pathogens used in this study. See the Additional file 3 caption for a detailed description (TXT 6 kb).
Additional file 5:
Phylogenetic tree of insect pathogens and non-pathogens used in this study. See the Additional file 3 caption for a detailed description (TXT 6 kb).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Takemoto, K., Aie, K. Limitations of a metabolic network-based reverse ecology method for inferring host–pathogen interactions. BMC Bioinformatics 18, 278 (2017). https://doi.org/10.1186/s12859-017-1696-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-017-1696-7