
Abstract
Background
The SYNGAP1 gene encodes for a small GTPase-regulating protein critical to dendritic spine maturation and synaptic plasticity. Mutations have recently been identified to cause a breadth of neurodevelopmental disorders including autism, intellectual disability, and epilepsy. The purpose of this work is to define the phenotypic spectrum of SYNGAP1 gene mutations and identify potential biomarkers of clinical severity and developmental progression.
Methods
A retrospective clinical data analysis of individuals with SYNGAP1 mutations was conducted. Data included genetic diagnosis, clinical history and examinations, neurophysiologic data, neuroimaging, and serial neurodevelopmental/behavioral assessments. All patients were seen longitudinally within a 6-year period; data analysis was completed on June 30, 2018. Records for all individuals diagnosed with deleterious SYNGAP1 variants (by clinical sequencing or exome sequencing panels) were reviewed.
Results
Fifteen individuals (53% male) with seventeen unique SYNGAP1 mutations are reported. Mean age at genetic diagnosis was 65.9 months (28–174 months). All individuals had epilepsy, with atypical absence seizures being the most common semiology (60%). EEG abnormalities included intermittent rhythmic delta activity (60%), slow or absent posterior dominant rhythm (87%), and epileptiform activity (93%), with generalized discharges being more common than focal. Neuroimaging revealed nonspecific abnormalities (53%). Neurodevelopmental evaluation revealed impairment in all individuals, with gross motor function being the least affected. Autism spectrum disorder was diagnosed in 73% and aggression in 60% of cases. Analysis of biomarkers revealed a trend toward a moderate positive correlation between visual-perceptual/fine motor/adaptive skills and language development, with posterior dominant rhythm on electroencephalogram (EEG), independent of age. No other neurophysiology-development associations or correlations were identified.
Conclusions
A broad spectrum of neurologic and neurodevelopmental features are found with pathogenic variants of SYNGAP1. An abnormal posterior dominant rhythm on EEG correlated with abnormal developmental progression, providing a possible prognostic biomarker.
Similar content being viewed by others


Introduction
Neurodevelopmental disorders caused by mutations in single genes such as and fragile X syndrome, Phelan-McDermid syndrome, and Rett syndrome offer unique insight into the pathogenesis of these disorders. One common mechanism among many neurodevelopmental disorders which has been suggested is excitatory/inhibitory imbalance leading to developmental and behavioral phenotypes. Shank3 knockout mice, for example, modeling Phelan-McDermid syndrome demonstrated reduced spine density as well as decreased miniature excitatory postsynaptic current frequency [1]. In a separate example, deleting Mecp2 in GABAergic neurons in mice revealed phenotypic similarities to whole-body deletion in mice modeling Rett syndrome suggesting a central role for inhibitory neuronal dysfunction [2]. Finally, Fmr1 knockout mice demonstrated reduced Gad67 levels in neuronal subsets, indicating reduced GABA production might underlie some of the phenotypes associated with fragile X syndrome [3]. Overall, these data indicate that single-gene mutations can lead to neurodevelopmental disorders through alteration in neuronal excitability.
One recently described single-gene disorder that results in excitatory/inhibitory imbalance is due to de novo loss-of-function mutations in Synaptic Ras GTPase-activating protein 1 gene (SYNGAP1, [4]). The SynGAP protein has a critical role in dendritic spine maturation and synaptic plasticity as described in mouse models of SYNGAP1 deficiency [5,6,7]. Localized to dendritic spines in neocortical pyramidal neurons, SynGAP has been found to have a role in NMDAR (N-methyl-d-aspartate receptor) activity and AMPAR (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor) trafficking [7, 8]. When NMDARs are activated by glutamate, Ca2+ ions enter the postsynaptic space, activating calmodulin-dependent protein kinase II (CaMKII) via phosphorylation and is maintained active via autophosphorylation. CaMKII then activates SynGAP, leading to endocytosis of AMPARs [9]. Heterozygous loss-of-function variants in SYNGAP1 result in reduced inhibition of the Ras pathway causing AMPAR exocytosis to the postsynaptic membrane [10]. Therefore, AMPAR exocytosis is increased which causes an excitatory/inhibitory imbalance, potentially leading to abnormalities during development.
Heterozygous deficiency of Syngap1 was shown to increase synaptic neurotransmission in mice and in cultured neurons treated with siRNAs [11]. Conversely, overexpression of SYNGAP1 in cultured neurons demonstrated a significant reduction in AMPAR-mediated miniature excitatory postsynaptic currents (mEPSCs), AMPAR surface expression, and AMPAR membrane insertion [11]. SynGAP also has multiple isoforms, which exert opposing effects on synaptic function. The α1 isoform has been shown to decrease mEPSC amplitude and frequency, while the α2 isoform increases mEPSC amplitude and frequency [12].
Loss-of-function variants in SYNGAP1 have been identified in individuals from cohorts with intellectual disability (syndromic and non-syndromic), autism, and epileptic encephalopathy [4, 13,14,15]. This suggests a tremendous breadth of clinical presentations for individuals with pathological mutations in SYNGAP1. Previous studies have reported individuals with SYNGAP1 deleterious variants and associated phenotypes. Reported phenotypic traits have included nearly universal epilepsy and intellectual disability/developmental delay, as well as variable presence of autism spectrum disorder and physical dysmorphisms [16, 17]. However, in-depth and longitudinal clinical characterization of a cohort of individuals ascertained for pathologic SYNGAP1 variants has not been undertaken. In this study, we present a cohort of patients with novel SYNGAP1 variants as well as patients with previously reported variants that expands our knowledge of pathogenic SYNGAP1 variants. We sought to comprehensively characterize the longitudinal clinical phenotypes in this population to determine the evolving spectrum of neurologic and neurodevelopmental abnormalities. Finally, in analyzing these data, we sought to identify potential elements within the serial neurologic and neurodevelopmental evaluations that could serve as biomarkers for disease identification, progression, and prognosis.
Methods
This study presents a retrospective review of clinical data from patients with pathogenic SYNGAP1 variants identified by clinical next-generation sequencing. All participating subjects were identified within a dedicated Primary Synaptopathy clinic at Texas Children’s Hospital in Houston, Texas. Patients were evaluated on one or several occasions over a 6-year period. Any patient with a deleterious SYNGAP1 variant was eligible to participate. Patients were excluded if additional potentially pathogenic variants in other genes with known association with neurodevelopmental disorders were also present.
Written informed consent was obtained from the parents of participants according to approved protocols by the Baylor College of Medicine Institutional Review Board.
Clinical data was retrospectively reviewed by a board-certified neurologist (JLH) and included personal and family history (including antenatal/perinatal history) and all history of care for seizures/epilepsy, development and behavior, physical examination, and neurologic examination. In addition, all historical neuroimaging (computed tomography, CT, or magnetic resonance imaging, MRI) was reviewed by pediatric neuroradiologists at our institution. All scalp electroencephalograms with a minimum of 21 electrode recordings in a standard 10–20 distribution were reviewed by board-certified neurophysiologists (RC and MQ). As an exploratory function, all traits within the standard clinical read were collected (background continuity, posterior dominant rhythm, symmetry and synchrony, interictal epileptiform discharges, ictal discharges, and other abnormalities not associated to epileptogenesis).
A neurodevelopmental evaluation was performed on all patients as part of the clinical evaluation. All neurodevelopmental data was reviewed by board-certified neurodevelopmental pediatricians (SV and SR). Whenever possible, clinical information was obtained with standardized testing by utilizing Gesell development schedules for gross motor (GM) assessment and The Capute Scales for infant development for visual-perceptual/fine motor (VP/FM) assessment and language/speech (LANG) assessment. The Capute Scales utilize two domains of assessment for this purpose: a Cognitive Adaptive Test that provides independent developmental quotients for VP/FM skills and can be extrapolated to evaluate for early non-verbal problem-solving skills and early adaptive skills and a Clinical Linguistic and Auditory Milestones Scale, which assesses early life receptive and expressive language. In addition, the combination of scores from The Capute Scales provides a Full-Scale Developmental Quotient (FSDQ) [18]. Whenever no standardized testing was formally obtained, clinical data was assessed to identify and determine a specific developmental quotient (DQ) at a minimum for the three domains described previously. In addition, we specifically evaluated key developmental milestones in patients harboring deleterious SYNGAP1 variants: ages for sitting unaided, walking independently, saying first word, scribbling, and using utensils by parental report.
Descriptive statistics were obtained for all variables, and data was classified into categorical and ordinal variables whenever relevant. Given the nature of this retrospective study, a variable number of time-points were available for analysis in this cohort. Data from patients in whom multiple data collection time-points were available (corresponding to follow-up visits at least 1 year apart) was utilized as independent data (i.e., in isolation) for the purpose of inferential data analysis. Select variables were tested for potential associations (by Fisher’s exact test) and correlations (by Pearson’s correlations) to determine possible biomarkers of clinical utility in assessing severity, progression, or prognosis. Normal distribution of all data was confirmed using the Shapiro-Wilk test for normalcy.
Results
A total of 15 subjects were identified in the study period, of which eight (53%) were male and seven (47%) were female. Mean age at diagnosis was 65.9 months (range 28–174 months).
Genetics
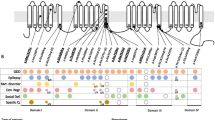
Fifteen individuals with seventeen SYNGAP1 variants which were discovered via clinical sequencing panels or exome sequencing are presented (Fig. 1, Table 1 and Additional file 1: Table S1). We compiled our data set, previously published studies, and likely pathogenic or pathogenic variants in SYNGAP1 found in ClinVar (Additional file 1: Table S1). From this combined dataset, there are seventeen variants which have been reported in our cohort or in previous publications that have been reported in ClinVar. Based on the date of submission in ClinVar compared to the date of publication for the corresponding paper, sixteen of these variants appear to be unique cases instead of repeated reports, with some variants being reported on multiple occasions in ClinVar. These data suggest that there might be loci within the SYNGAP1 gene that are prone to mutation. For example, one variant (p.Pro562Leu) reported in Mignot et al. [16] has been reported four times in ClinVar and lies within the RASGAP domain of SynGAP. Additionally, another variant (p.Arg143*) reported in two publications [15, 16] was also reported four times in ClinVar.

Pathogenic variants in SYNGAP1. a Diagram of SYNGAP1 gene. Pathogenic variants reported in this study above the gene. Previously published pathogenic variants below the gene. Repeat variants between this study and previously published variants represent separate, unique individuals to the best of our knowledge. b Diagram of SynGAP protein. Pathogenic protein variants from this study and ClinVar above the protein. Previously published protein variants below the protein. Repeat variants between this study and previously published variants represent separate, unique individuals to the best of our knowledge
We then compared this compiled dataset of pathogenic variants to the Exome Aggregation Consortium (ExAC) database of control exomes from individuals without neurodevelopmental disorders [19]. None of these variants were represented in the ExAC database. Furthermore, SYNGAP1 has a probability of loss of function intolerance (pLI) of 1.0 in ExAC, demonstrating a high probability that LoF variants cause a severe phenotype precluding inheritance of deleterious variants. The Z score of deviation from expected allelic frequency for missense mutations in SYNGAP1 is 7.15 (539.6 expected variants, 200 observed), again demonstrating intolerance for deleterious variants. We also examined the gnomAD database to determine if any of our patient variants were in this database of 123,136 exomes and 15,496 genomes from neurotypical individuals. One variant (p.Ser898Cys) was present in both gnomAD and a patient from our cohort as well as being predicted as deleterious through algorithms CADD, PolyPhen2, and SIFT. However, this patient had two additional variants in SYNGAP1 (p.Glu582*, p.Leu769Val), the former of which we believe to be causative.
We investigated the pathogenicity of missense variants in our data set and in previously published data sets by using predictive algorithms CADD, PolyPhen2, PROVEAN, and SIFT. In CADD, each variant is assigned an evolutionary action (EA) score which correlates with loss of protein function [20]. In the combined missense variant data set from all sources (ClinVar, previous publications, and our data), thirteen out of twenty variants had an EA score close to or above 80, strongly suggesting deleterious impact on protein function (Additional file 1: Table S1). In PolyPhen2, eighteen out of twenty missense variants were predicted to be “probably damaging” with the other two variants predicted to be “possibly damaging.” PROVEAN predicted all but four missense variants as being “deleterious,” and SIFT predicted all but two missense variants as being “damaging.” These missense variants were observed throughout SynGAP, three in the PH domain, two in the C2 domain, nine in the RASGAP domain, and the remaining six variants interspersed throughout the protein.
Neurophysiologic manifestations
At least one scalp EEG was performed on all patients. Epileptiform activity was captured in fourteen of fifteen individuals with generalized discharges being more common than focal (Table 1). Occipital epileptiform discharges were substantially more common than from other areas (Fig. 2a, e). The epileptiform discharges took the form of polyspikes as well as spike and slow wave (Fig. 2b). Epileptiform activity was potentiated in five children with sleep onset. Slowing or absence of the posterior dominant rhythm (relative to chronologic age) was also a common manifestation on electroencephalograms (12 of 15). Only three out of eight individuals greater than 5 years old achieved an alpha rhythm (8 Hz or greater) on EEG. In addition, nine of our fifteen patients displayed intermittent rhythmic delta activity (IRDA), with the majority of those (6 of 9) having occipital predominance (Fig. 2c).

Electrographic and epileptic features in patients with SYNGAP1 pathogenic variants. a Example electroencephalogram containing occipital spikes from patient with pathogenic SYNGAP1 variant. b Example electroencephalogram containing polyspikes from patient with pathogenic SYNGAP1 variant. c Example electroencephalogram containing rhythmic delta waves from patient with pathogenic SYNGAP1 variant. d Example electroencephalogram containing seizure activity from patient with pathogenic SYNGAP1 variant. Patient’s seizure was characterized by behavioral arrest. e Quantification of interictal epileptiform activity in patients with SYNGAP1 pathogenic variants
Epilepsy
All fifteen individuals in our cohort with germline SYNGAP1 pathogenic variants were diagnosed with epilepsy. The mean age of onset for seizures was 38 months ± 32 (SD) with a range of 12 to 120 months. The most common seizure semiology was atypical absence (9 of 15) (example in Fig. 2d), followed by absence (4 of 15), then generalized tonic-clonic and atonic (three each), and finally two patients with focal onset seizures.
Developmental progression
Developmental testing data was obtained for all individuals (Additional file 2: Table S2). Standardized testing for gross motor development was performed for eight of our fifteen children with deleterious germline SYNGAP1 mutations using the Gesell development schedules [21]. Significant delay was noted in all individuals. Three individuals had repeated testing over yearly (or greater) intervals. For all of these individuals, there was plateauing of development at approximately 60 months of age. Overall there was only a suggestion of a mild correlation between chronological age and age equivalents for gross motor development of this cohort (Fig. 3a), while there was a moderate negative correlation between gross motor developmental quotient and chronological age (Fig. 3b); however, neither reached statistical significance.

Developmental correlations with chronological age. a Gross motor age equivalents plotted against chronological age. b Gross motor developmental quotient (DQ) plotted against chronological age. c Fine motor age equivalents plotted against chronological age. d Fine motor DQ plotted against chronological age. e Language age equivalents plotted against chronological age. f Language DQ plotted against chronological age. g Age at which select developmental milestones occur in SYNGAP1 patient cohort. Mean ± standard deviation
Visual-perceptual/fine motor skill (VP/FM) was evaluated by the Cognitive Adaptive Test (CAT) component of The Capute Scales [18] for ten subjects with pathogenic SYNGAP1 variants on at least one occasion. Similar to gross motor skills, VP/FM skills only demonstrated a mild positive correlation that was not statistically significant between age equivalents and chronological age (Fig. 3c), while also demonstrating a trend toward mild to moderate negative correlation between developmental quotient and chronological age (p = 0.1) (Fig. 3d).
Language development was evaluated for ten subjects with pathogenic SYNGAP1 variants using the Clinical Linguistic and Auditory Milestone Scale (CLAMS) component of The Capute Scales [18]. In contrast to gross motor and VP/FM development, language skills trended toward a moderate positive correlation between age equivalence and chronological age (p = 0.07) (Fig. 3e). There was a mild trend toward a negative correlation between language developmental quotient and chronological age (p = 0.142) (Fig. 3f).
We specifically evaluated the timing of acquisition of key developmental milestones in patients harboring deleterious SYNGAP1 variants: ages for sitting unaided, walking independently, saying first word, scribbling spontaneously, and using utensils. All of these were found to be significantly delayed in our cohort of children (Fig. 3g).
When evaluating neurophysiologic data and development, only VP/FM (p = 0.1) and language skills (p = 0.114) displayed a trend toward moderate correlation between age equivalents and posterior dominant frequency (Fig. 4a–c). The frequency of the posterior dominant rhythm (PDR) itself did not correlate with age in its expected chronologic-developmental maturation; this suggests that the uncovered development-PDR correlations were not simply due to changes in PDR with age (Fig. 4d). There was no association between the presence of IRDA and severity of developmental disability in any domain (mild/moderate vs. severe/profound disability, p = 0.235).

Developmental correlations with posterior dominant rhythm (PDR). a Gross motor age equivalents plotted against posterior dominant rhythm (PDR). b Fine motor age equivalents plotted against PDR. c Language age equivalents plotted against PDR. d PDR plotted against chronological age
Other neurologic manifestations
Strabismus was present in over 60% of our patients with over 60% of those patients requiring corrective surgery. Sleep abnormalities were reported in two thirds of our patients with insomnia manifested as nighttime awakenings being the predominant issue. Only one patient was identified as having obstructive sleep apnea. This suggests that for most patients, the insomnia was primary. Neurologic examination revealed low muscle tone in one third of our patients and elevated tone in only one. While general motor strength was normal in these patients, significant ataxia was identified in 21%.
Neuroimaging characteristics
Magnetic resonance imaging (MRI) of the brain was obtained in all patients in our cohort at an average age of 44.6 months ± 30 months (SD). Seven patients (47%) had normal neuroimaging. Among those with an abnormal MRI, nonspecific white matter hyperintensities were observed on T2/FLAIR sequences in four patients with variable location including frontal lobes, centrum semiovale, subinsular, and periatrial. One patient had an isolated Chiari I malformation, whereas another had Chiari I and a thickened corpus callosum. A predominantly frontal, mildly simplified gyral pattern was present in one patient, and another had several small developmental venous anomalies (Additional file 3: Figure S1).
Neurobehavioral manifestations
Behavioral abnormalities were amply described in our cohort. Aggressive behavior toward caregivers and siblings was reported in 60% of our patients at any point in time. Self-injurious behavior of any nature was reported in a third of our cohort, described most frequently as biting oneself. Disruptive hyperactivity was also reported in a third of our patients. Eleven of fifteen (73%) have been diagnosed with autism spectrum disorder by a medical provider.
Discussion
Deleterious variants in SYNGAP1 have been identified in a variety of phenotypically defined cohorts including syndromic/non-syndromic intellectual disability, autism spectrum disorder, and epileptic encephalopathy [4, 6, 13, 15, 16]. In our cohort of children with pathogenic SYNGAP1 variants, we corroborated developmental delay/intellectual disability and epilepsy as universal features.
Neurodevelopmental traits-global delay/disability must be analyzed considering the evolving nature of developmental test scoring. The universality and severity of neurocognitive impairment is presented in Table 1 and is derived from the assessment of developmental quotients (DQ = age equivalence/chronologic age), with severity of impairment most significant in language skills and least in gross motor skills. Our data is similar to prior cohorts in the preponderance of severe disability, especially evident at a later age [15, 16]. Our cohort displayed weak to moderate positive correlations between the developmental and chronologic age (Fig. 3a, c, e), contrasted with a weak to moderate negative correlation between developmental quotients and chronologic age (Fig. 3b, d, f). We believe this to be expected given the growing dissociation between chronologic and developmental age, without of a true plateau or regression, in development. It may be most useful in future clinical studies to utilize age equivalents in measuring developmental progression in these individuals.
Diagnosis of autism spectrum disorder was present in just under three quarters of our cohort, comparable with prior reports ranging from 50 to 80% [15, 16]. It has been proposed that while mutations in the SYNGAP1 gene likely play a role in the pathogenesis of ASD, their presence is potentially not sufficient for its development [16]. In addition, the variable manifestations of ASD can evolve with age, suggesting that an absence of this diagnosis at a single point in time may not hold true later, and ASD may increase in prevalence with age in this population.
Prior studies have linked neurophysiologic abnormalities to developmental outcomes such as in epileptic encephalopathies and autism spectrum disorders (ASDs) [16]. Capal et al. recently suggested abnormal EEG patterns, even in the absence of epilepsy, were linked with worse developmental outcomes in children with ASD [22]. Similarly, abnormal EEG patterns have been linked to a number of other genetically defined intellectual and developmental disorders [23,24,25]. Based on our data, we hypothesize this might be true for patients with SYNGAP1 mutations. First, most children in our study were found to have interictal epileptiform discharges. For the majority of our subjects, there was a posterior prominence of these discharges. These discharges were enhanced with sleep in a subset of our patients. Second, the majority of our subjects also displayed intermittent rhythmic delta activity. Third, we identified abnormally slow posterior dominant rhythm for age in the majority of our patients. The frequency of the PDR is known to increase with development typically achieving the alpha range (8–12 Hz) by 4–5 years of age. Only three of fifteen individuals in our cohort achieved an alpha frequency of their PDR despite most having an EEG after their fourth birthday (11 of 15).
One of the greatest challenges for developing targeted therapies for neurodevelopmental disorders is in identifying quantitative biomarkers that directly correspond to clinical outcomes. Indeed, lack of such biomarkers for most neurodevelopmental disorders has potentially led to disappointing results for late-stage clinical trials such as for fragile X syndrome [26]. As described above, we have identified several neurophysiologic features that warrant further investigation as potential biomarkers for disease progression identified in our cohort. The most salient findings include a moderate correlation between developmental age equivalence in language and VP/FM development with the frequency of the posterior dominant rhythm. This correlation is not simply due to maturation of the posterior dominant rhythm with age as most of our data comes after 4 years of age when the PDR has achieved maturity. Furthermore, plotting PDR frequency versus chronological age in our cohort confirmed that there was no correlation. Despite the listed descriptions of EEG anomalies in other neurodevelopmental conditions, there has not been—to our knowledge—a prior report of a correlation with PDR frequency and developmental progression for any genetically defined disorder.
Some trends began to emerge in the genetic architecture of pathogenic variants in SYNGAP1. First, combining our data with all previously published mutations and pathogenic variants from ClinVar, the majority of mutations fall within exons 3–17, sparing the first two (except for a single exon 1 mutation) and last two exons. Why these exons are spared is unclear but might be due in part to the extensive alternative slicing that occurs in the five prime and three prime regions of the SYNGAP1 transcript. Second, while we have identified a small number of recurrent mutations in SYNGAP1, the vast majority are novel non-sense or frameshift variants confirming that patients’ variants must largely be loss of function.
Our study has multiple limitations. Since this was a retrospective study, we have performed an exhaustive data analysis to the extent permitted by clinical documentation within a specialty clinic. This has limited the number of potential time-points in a single individual that can be analyzed for all desired parameters, to establish comprehensive neurodevelopmental and neurobehavioral trajectories. Given the overall neurologic stability, patients are seldom followed more frequently than every 6 months, and we deliberately collected only time-points at least 1 year apart to allow for developmental changes to clearly emerge. Inference and conclusions regarding neurodevelopmental trajectories (progression, regression, or plateauing) are limited. Thus, these data are exploratory and hypothesis generating. Future collection in a prospective observational manner may better help standardize and enrich these findings. Given the rarity of genetically confirmed SYNGAP1 patients, the size of our current cohort also limits potential inferences, including describing a clear genotype-phenotype correlation with any of the developmental parameters evaluated in these patients.
Conclusions
Evaluation of neurodevelopmental progression in individuals with pathogenic SYNGAP1 variants revealed a broad spectrum, most often ranging from moderate to severe impairment. Furthermore, the frequency of posterior dominant rhythm of EEG in these individuals revealed a trend toward correlation with developmental progression, providing a possible prognostic biomarker. Further assessment using more extensive neuropsychological cognitive/developmental evaluation in a prospective manner with larger cohorts will better define developmental progression and provide benchmarks for future targeted clinical trials for individuals with pathogenic SYNGAP1 variants. In this regard—and given the rarity of this genetically defined neurodevelopmental disorder—expanding the number of participants in these cohorts may require inter-institutional collaborations to achieve larger sample sizes and potentially statistical significance.
Availability of data and materials
The data sets used and/or analyzed in the current study are available from the corresponding author on reasonable request.
Abbreviations
- AMPAR:
-
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- CaMKII:
-
Calmodulin-dependent protein kinase II
- CAT:
-
Cognitive Adaptive Test (part of The Capute Scales)
- CLAMS:
-
Clinical Linguistic and Auditory Milestone Scale (part of The Capute Scales)
- EEG:
-
Electroencephalogram
- IRDA:
-
Intermittent Rhythmical Delta Activity
- mEPSC:
-
Miniature excitatory postsynaptic currents
- MRI:
-
Magnetic resonance imaging
- NMDAr:
-
N-Methyl-d-aspartate receptor
- PDR:
-
Posterior dominant rhythm
- VNS:
-
Vagal nerve stimulator
References
Mei Y, Monteiro P, Zhou Y, Kim JA, Gao X, Fu Z, et al. Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature. 2016;530(7591):481–4.
Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468(7321):263–9.
Ruby K, Falvey K, Kulesza RJ. Abnormal neuronal morphology and neurochemistry in the auditory brainstem of Fmr1 knockout rats. Neuroscience. 2015;303:285–98.
Hamdan FF, Gauthier J, Spiegelman D, Noreau A, Yang Y, Pellerin S, et al. Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N Engl J Med. 2009;360(6):599–605.
Vazquez LE, Chen HJ, Sokolova I, Knuesel I, Kennedy MB. SynGAP regulates spine formation. J Neurosci. 2004;24(40):8862–72.
Clement JP, Aceti M, Creson TK, Ozkan ED, Shi Y, Reish NJ, et al. Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell. 2012;151(4):709–23.
Kim JH, Lee HK, Takamiya K, Huganir RL. The role of synaptic GTPase-activating protein in neuronal development and synaptic plasticity. J Neurosci. 2003;23(4):1119–24.
Kim MJ, Dunah AW, Wang YT, Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46(5):745–60.
Oh JS, Manzerra P, Kennedy MB. Regulation of the neuron-specific Ras GTPase-activating protein, synGAP, by Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 2004;279(17):17980–8.
Krapivinsky G, Medina I, Krapivinsky L, Gapon S, Clapham DE. SynGAP-MUPP1-CaMKII synaptic complexes regulate p38 MAP kinase activity and NMDA receptor-dependent synaptic AMPA receptor potentiation. Neuron. 2004;43(4):563–74.
Rumbaugh G, Adams JP, Kim JH, Huganir RL. SynGAP regulates synaptic strength and mitogen-activated protein kinases in cultured neurons. Proc Natl Acad Sci U S A. 2006;103(12):4344–51.
McMahon AC, Barnett MW, O'Leary TS, Stoney PN, Collins MO, Papadia S, et al. SynGAP isoforms exert opposing effects on synaptic strength. Nat Commun. 2012;3:900.
Berryer MH, Hamdan FF, Klitten LL, Moller RS, Carmant L, Schwartzentruber J, et al. Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum Mutat. 2013;34(2):385–94.
Parker MJ, Fryer AE, Shears DJ, Lachlan KL, McKee SA, Magee AC, et al. De novo, heterozygous, loss-of-function mutations in SYNGAP1 cause a syndromic form of intellectual disability. Am J Med Genet A. 2015;167A(10):2231–7.
Carvill GL, Heavin SB, Yendle SC, McMahon JM, O'Roak BJ, Cook J, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013;45(7):825–30.
Mignot C, von Stulpnagel C, Nava C, Ville D, Sanlaville D, Lesca G, et al. Genetic and neurodevelopmental spectrum of SYNGAP1-associated intellectual disability and epilepsy. J Med Genet. 2016;53(8):511–22.
Kimura Y, Akahira-Azuma M, Harada N, Enomoto Y, Tsurusaki Y, Kurosawa K. Novel SYNGAP1 variant in a patient with intellectual disability and distinctive dysmorphisms. Congenit Anom (Kyoto). 2018;58(6):188–90.
Kube DA, Wilson WM, Petersen MC, Palmer FB. CAT/CLAMS: its use in detecting early childhood cognitive impairment. Pediatr Neurol. 2000;23(3):208–15.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91.
Katsonis P, Lichtarge O. A formal perturbation equation between genotype and phenotype determines the Evolutionary Action of protein-coding variations on fitness. Genome Res. 2014;24(12):2050–8.
Ball RS. The Gesell developmental schedules: Arnold Gesell (1880-1961). J Abnorm Child Psychol. 1977;5(3):233–9.
Capal JK, Carosella C, Corbin E, Horn PS, Caine R, Manning-Courtney P. EEG endophenotypes in autism spectrum disorder. Epilepsy Behav. 2018;88:341–8.
Alfei E, Raviglione F, Franceschetti S, D'Arrigo S, Milani D, Selicorni A, et al. Seizures and EEG features in 74 patients with genetic-dysmorphic syndromes. Am J Med Genet A. 2014;164A(12):3154–61.
Heard TT, Ramgopal S, Picker J, Lincoln SA, Rotenberg A, Kothare SV. EEG abnormalities and seizures in genetically diagnosed Fragile X syndrome. Int J Dev Neurosci. 2014;38:155–60.
Asadi-Pooya AA. Yield of EEG monitoring in children with developmental disabilities is high. Epilepsy Behav. 2018;84:105–6.
Duy PQ, Budimirovic DB. Fragile X syndrome: lessons learned from the most translated neurodevelopmental disorder in clinical trials. Transl Neurosci. 2017;8:7–8.
Acknowledgements
We are indebted to the patients and their families for participating in this study.
Funding
HHS-NIH National Institute of Neurological Disorders and Stroke (NINDS) Valid: J. Lloyd Holder NS091381; Doris Duke Charitable Foundation (DDCF) Valid: J. Lloyd Holder 2017063.
Author information
Authors and Affiliations
Contributions
AJ-G contributed to the data collection, clinical data analysis, and manuscript drafting and revision. SN contributed to the data collection, genetic data analysis, and manuscript drafting and revision. FA-P contributed to the data collection, genetic data analysis, and manuscript revision. EAM and AB contributed to the data collection, clinical data analysis, and manuscript revision. DH contributed to the clinical data analysis and manuscript revision. RC and MQ contributed to the clinical data collection, EEG review and analysis, and manuscript revision. SV and SR contributed to the neurodevelopmental data analysis and manuscript revision. JLH contributed to the project design, data collection and analysis, and manuscript drafting and revision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Institutional Review Board at Baylor College of Medicine. Written informed consent to participate was obtained from the guardians of all participating subjects.
Consent for publication
Consent for publication of individual EEG and MRI images was obtained from guardians of all participating subjects.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Compiled list of all pathogenic variants. Table contains all pathogenic variants in the current report, previously published, and from the ClinVar database. (XLSX 18 kb)
Additional file 2:
Table S2. Developmental and behavioral phenotypes of a SYNGAP1 cohort. Table contains developmental and behavioral findings for each individual including clinically relevant milestones in each of gross motor, visual-perceptual/fine motor, and language domains, as well as neuropsychiatric findings including autism spectrum disorder. (DOCX 15 kb)
Additional file 3:
Figure S1. MRI imaging findings. (A) Subject #1 at 34 months. Sagittal T1 (left) and coronal T2 (right) images show diffuse mildly simplified gyri, predominantly in the frontal lobes. (B) Subject #13 at 6 years and 6 months. Axial T2/FLAIR images show diffuse hyperintense signal in the bilateral centrum semiovale (left, arrows) and punctate foci of subcortical white matter hyperintensity (right, arrowheads). (C) Subject #10 at 11 years and 4 months. Sagittal T2/FLAIR images demonstrate diffusely thickened corpus callosum (arrows) and a mild Chiari I malformation (arrowheads). (PPTX 242 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jimenez-Gomez, A., Niu, S., Andujar-Perez, F. et al. Phenotypic characterization of individuals with SYNGAP1 pathogenic variants reveals a potential correlation between posterior dominant rhythm and developmental progression. J Neurodevelop Disord 11, 18 (2019). https://doi.org/10.1186/s11689-019-9276-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11689-019-9276-y