
Abstract
Background
A new sensitive colorimetric determination of nifedipine has been developed following azo dye formation with 4-carboxyl-2,6-dinitrobenzenediazonium ion (CDNBD). Judging from the various generic brands currently now available, this research was conceived as a means of developing an alternative cost-effective and readily adaptable method for the assay of nifedipine in tablets and for which official high performance liquid chromatographic technique may not be readily available.
Results
Nifedipine was reduced with Zn/HCl reduction system and then the diazo coupling reaction was carried out with the CDNBD reagent to generate a new azo adduct with optimal wavelength at 470 nm representing a bathochromic shift relative to nifedipine, reduced nifedipine and CDNBD reagent. Optimal temperature and time for coupling were selected as 50 oC and 15 minutes. A linear response was observed over 2.9 -14.5 µg/mL of nifedipine with a correlation coefficient of 0.9985 and the drug combined with CDNBD at a stoichiometric ratio of 2:1. The method has limits of detection and quantitation of 0.1344 µg mL-1 and 0.4074 µg mL-1 respectively. The Sandell’s sensitivity obtained is 4.673 ng/cm2 and the method was reproducible over a three day assessment. Intra- and inter-day accuracies (in terms of errors) were of the order -0.008 to 3.22 % while precisions were generally less than 3.1 % (RSD).
Conclusions
The developed spectrophotometric method is of equivalent accuracy (p > 0.05) with USP 2007 HPLC method. It has the advantages of speed, simplicity, sensitivity and more affordable instrumentation and could find application as a rapid and sensitive analytical method for nifedipine. It is the first described method by azo dye derivatization for the analysis of nifedipine in bulk samples and dosage forms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Nifedipine, chemically dimethyl-2, 6-dimethyl-4-(2-nitrophenyl)-1, 4-dihydropyridine-3, 5-dicarboxylate, has an empirical formula of C17H18N2O6. It is commonly available in capsule and extended-release tablet dosage forms. Nifedipine, which occurs as a yellow crystalline powder, has a melting point of 172°C to 174°C and is practically insoluble in water, sparingly soluble in dehydrated alcohol and freely soluble in acetone [1–3]. When exposed to daylight or to certain wavelengths of artificial light, it is converted to a nitrosophenylpyridine derivative, and exposure to ultraviolet light leads to the formation of the same derivative [1]. The drug is official in the United States Pharmacopoeia (USP) [4], which recommends high performance liquid chromatography (HPLC) for its assay (both the pure drug and its dosage forms), and in the BP [3], which recommends redox titration using cerium sulphate and HPLC for the assay of the drug and its dosage forms, respectively.
Nifedipine, the prototypical 1,4-dihydropyridine, is a calcium channel blocker with peripheral and coronary vasodilator activity. It is used in the management of hypertension, angina and some other cardiovascular disorders. Its uses result primarily in vasodilatation, with reduced peripheral resistance, blood pressure, and afterload; increased coronary blood flow; and a reflex increase in heart rate. This in turn results in an increase in myocardial oxygen supply and cardiac output. It acts by inhibiting the transmembrane influx of calcium into cardiac and vascular smooth muscle cells. Nifedipine is rapidly and almost completely absorbed from the gastrointestinal tract but undergoes extensive hepatic first-pass metabolism. Bioavailability of oral capsules is between 45% and 75% but is lower for longer-acting formulations. Peak blood concentrations were reported to occur 30 min after oral doses of capsules. It is about 92% to 98% bound to plasma proteins and is distributed into breast milk. It is extensively metabolised in the liver, and 70% to 80% of a dose is excreted in the urine almost entirely as inactive metabolites. The half-life is about 2 h after intravenous doses or oral capsules [1].
Detailed survey of literature for nifedipine revealed several methods that have been reported for the assay of nifedipine either alone or in combined form in drug formulations. These analytical techniques include UV-Visible (Vis) spectrophotometry [5–20], HPLC [21–28], high performance thin layer chromatography [29], micellar electrokinetic chromatography [30], electroanalytical methods [31–33], flow injection analysis [34] and mass spectrometry [35].
Nifedipine alone has been estimated by second derivative UV spectrophotometry [8], whereas first derivative UV spectrophotometry was utilised for its assay in combined dosage forms [9, 10]. Two kinetic spectrophotometric methods have been reported: One is based on the oxidation of the drug with KMnO4 at neutral pH [11], and the other method is based on the reduction of iron(III) by the drug and subsequent interaction of iron(II) with ferricyanide to form Prussian blue [12]. Some other spectrophotometric methods involve a reduction of the nitro group of nifedipine with Zn/NH4Cl and Zn/HCl to hydroxylamino and primary aromatic amino derivatives, respectively. The hydroxylamino derivative was reacted with 4-(methylamino) phenol and potassium dichromate [13], while the primary aromatic amino derivative formed Schiff's bases with 3,4,5-trimethoxybenzaldehyde [14] and 4-dimethylaminobenzaldehyde [15]. Determinations of the drug in pharmaceutical preparations via extractive spectrophotometric methods have also been reported [7, 16]. Other reported methods include the following: one involving a reaction of the nitro group of the drug with potassium hydroxide in dimethyl sulphoxide [17], the oxidation of the drug with ammonium molybdate [17], the formation of blue-coloured complex with Folin Ciocalteau reagent [18] and another based on the formation of a charge transfer complex with chloranil [19]. Recently, Revathi et al. reported a spectroscopic method based on hydrotropic solubilisation in which the determination of nifedipine in tablet formulations was done using 40% sodium salicylate solution [20]. Majority of these methods suffer from the disadvantage of extensive extraction procedures, long analytical time and high calibration range.
In continuation of our previous efforts in the analysis of organic compounds of pharmaceutical importance through azo dye chemical derivatization with 4-carboxyl-2,6-dinitrobenzene diazonium (CDNBD) ion [36–44], we report in this paper a new simple, accurate and precise colorimetric method for the assay of nifedipine in bulk samples and tablet dosage forms.
Methods
Preparation of reduced nifedipine stock solution
A quantity of nifedipine CRS crystals (11.0 mg) was weighed and dissolved in 1 mL of glacial acetic acid in a 20-mL beaker. This was warmed gently to ensure complete dissolution. Two millilitres of concentrated HCl was added followed by 400 mg of Zn dust. The whole content was allowed to stand for 5 min. The solution was then filtered through a cotton wool plug into a 10-mL volumetric flask and made up to volume by adding fresh glacial acetic acid rinsing the filter aid in the process. This gave a 1 mg/mL of reduced nifedipine (RN) solution. A stock solution of 100 μg/mL was thereafter made from this and kept in a wrapped sample vial.
Preparation of CDNBD solution
The reagent solution of CDNBD ion was prepared in concentrated sulphuric acid following diazotization of 4-amino-3,5-dinitrobenzoic acid with sodium nitrite as previously reported [45].
Selection of analytical wavelength
A 0.1-mL RN stock solution was added into a test tube containing 0.5 mL of CDNBD solution. The mixture was vortex mixed for 10 s, and the reaction tube was placed in a thermostated water bath maintained at 50°C for 15 min. The reaction mixture was thereafter placed in an ice bath; 2 mL of ice-cold distilled water was added and made up to 5 mL with acetonitrile. The formation of the azo adduct was monitored by normal-phase thin layer chromatography (TLC) using ethyl acetate-methanol mixture (9:1 and 8:2). The absorption spectrum of the new orange-coloured adduct was determined against CDNBD, nifedipine and RN blanks, using the UV–vis spectrophotometer. The analytical wavelength (λmax) was then determined by inspection of the overlaid spectra. The wavelength where the difference in absorptivity is maximal was selected as the analytical wavelength.
Optimisation studies
Temperature and time were optimised using the method of steepest ascent. A 0.1 mL of the RN stock solution was coupled with 0.5 mL of CDNBD in a test tube, and the reaction mixture was mixed using a vortex mixer. The sample tubes were incubated at 30°C and 50°C for 5 and 20 min. Similar experiments were carried out at 60°C and 80°C. At the end of each time interval, the reaction mixture was placed in an ice bath, and the reaction was terminated by adding 2 mL of ice-cold distilled water and making up to 5 mL with acetonitrile. The absorbance readings of the azo adduct were taken at 470 nm on the digital colorimeter. Each determination was done in duplicate.
The optimal reaction time was determined by studying the effect of varied time allowed for coupling to take place at 50°C between RN and CDNBD at 5, 10, 15, 20 and 30 min. A 0.1 mL of RN stock solution was added to 0.5 mL of the reagent in test tubes. The azo adduct formed was processed as described above. The optimal reaction time was taken as the time corresponding to maximal absorbance of the samples. Each determination was done in duplicate.
The effect of the diluting solvent on coupling reaction was investigated using acetonitrile, ethanol, methanol and 1-propanol. In each case, the reaction mixture was terminated by adding 2 mL of ice-cold distilled water and making up to 5 mL with the respective solvents. Absorbance readings were taken at 470 nm.
Determination of stoichiometric ratio for the formation of azo dye
Aliquots of CDNBD reagent, 0, 0.2, 0.25, 0.33, 0.5, 0.67, 0.75, 0.8 and 1.0 mL, were added respectively into nine test tubes. Each tube was in turn made up to 1.0 mL with the drug stock solution. The mixture was vortex mixed for 10 s followed by incubation at 50°C for 15 min. Afterwards, the reaction was terminated by adding 2 mL of ice-cold distilled water and making up to 5 mL with acetonitrile. A series of blank determinations were carried out in which the volume of the RN stock solution was replaced with glacial acetic acid. The absorbance was measured at 470 nm against the blank, and values obtained were plotted against mole fraction of the reagent solution. Each determination was carried out in duplicate.
Validation studies
The calibration curve for the azo adduct was prepared using a concentration range of 0 to 14.5 μg/mL from the stock of RN solution in glacial acetic acid. To seven test tubes, each containing 0.67 mL of CDNBD reagent solution, 0, 0.05, 0.075, 0.1, 0.15, 0.2 and 0.25 mL corresponding to 0, 2.88, 4.35, 5.8, 8.7, 11.6 and 14.5 μg/mL of RN stock solution were added respectively. The mixture was vortex mixed for 10 s and incubated at 50°C for 15 min. For each sample, the azo adduct formed was processed as described above; the absorbance reading was recorded at 470 nm. Calibration curves were prepared everyday for 3 days, and the average absorbance values were used in constructing the working calibration curve. The regression line equation and correlation coefficient were obtained from the calibration curve.
Accuracy and precision were determined through recovery studies. These were determined using quadruplicate samples at three concentration levels of 4.35, 8.7 and 11.6 μg/mL on three successive days. The volumes of RN solution corresponding to these concentrations were measured, i.e. 0.075, 0.15 and 0.25 mL. Each volume was added into test tubes containing 0.67 mL of CDNBD reagent solution. The reaction mixture was vortex mixed for 10 s and incubated at 50°C for 15 min. The azo adduct formed was processed as described above, and the absorbance reading was taken at 470 nm using acetonitrile as blank solvent. This was done four times for each volume. A 3-day recovery study was done, and the accuracy and precision were determined from the regression equation.
Application to tablet dosage form analysis
Six brands of nifedipine extended-release tablets (20 mg) were analysed using the new colorimetric method developed in this study. Uniformity of weight test was carried out on all the brands using 10 tablets.
The assay of active ingredients for the brands was determined based on the new and official methods. For the assay of the six brands of nifedipine tablets, six different analyte stock solutions (A to F) of the same concentration were prepared. Based on uniformity of weight test, 105 mg of Frapine® (Medrel Pharmaceutical Ltd., Mumbai, India), 113 mg of Nifedin Dexcel® (Dexcel Ltd., Hamilton, New Zealand), 47 mg of Nifegem® (Gemini Pharmaceuticals Ltd., Commack, NY, USA), 46 mg of Nifecard® (Salutas Pharma GmbH, Barleben, Germany), 45 mg of Medipine® (Cipla Ltd., Mumbai, India) and 75 mg of Nife-D® (Lincoln Pharmaceuticals Ltd., Ahmedabad, India) powdered tablets each corresponding to 11 mg of nifedipine were weighed and dispersed in 1.0 mL of glacial acetic acid in a 20-mL beaker. This was warmed gently to aid dissolution of the active ingredient. Two millilitres of concentrated HCl was added followed by 400 mg of Zn dust. The whole content was allowed to stand for 5 min. The solution was then filtered through a cotton wool plug into a 10-mL volumetric flask and made up to volume by adding fresh glacial acetic acid rinsing the filter aid in the process. This gave a 1 mg/mL of RN solution. An analyte stock solution of 100 μg/mL was thereafter made from this and kept in a wrapped sample vial.
From each analyte stock solution (A to F), 0.15 mL was transferred into six tubes in turn containing 0.67 mL of CDNBD reagent solution. The mixture was vortex mixed for 10 s and incubated at 50°C for 15 min. Afterwards, the tubes were transferred into an ice bath; 2 mL of ice-cold distilled water was added and made up to 5 mL with acetonitrile. The absorbance readings of the azo adduct were measured at 470 nm on the colorimeter.
The analysis of the tablet samples was also repeated using the USP [3] HPLC procedures, which were adopted as the official procedure.
Interference studies
A 0.15-mL quantity of the stock corresponding to 8.7 μg/mL of RN was spiked into tablet excipients containing 5 mg each of lactose, starch, gelatine, magnesium stearate, talc and a mixture of the five excipients. Coupling and processing of the azo adduct that was formed were carried out as described above.
Results and discussion
Chemistry of the reaction
The diazo coupling reaction between CDNBD reagent solution and RN gave an instant orange-yellow colour that became deeper with time. The colour formation indicates the formation of an azo dye (Scheme 1). This colour is distinct from the colourless solution of RN in glacial acetic acid. TLC analysis revealed the formation of a new compound. As shown in Scheme 1, the nitro group on nifedipine was reduced to an amino group by Zn/HCl. This is accompanied by the cleavage of the two ester linkages (to give carboxylic acids) since the reaction is taking place in an acidic medium at a slightly elevated temperature. Another possible reaction occurring in the medium is the aromatization of the dihydropyridine moiety to pyridine. This is so because aromatization is accomplished most easily in dihydropyridines and much more when the ring is fused to an aromatic ring [46] as is the case in nifedipine. The end result of all these reactions is the formation of new species which on coupling with CDNBD elongates the chromophoric conjugation, leading to absorption of light in the visible region.

Proposed coupling pattern between reduced nifedipine and CDNBD.
Selection of analytical wavelength
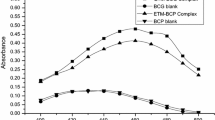
The absorption spectrum of nifedipine shows two absorption maxima at 230 nm and 333 nm, while RN shows two absorption maxima at 224 nm and 310 nm. The spectrum of the adduct shows two maxima at 230 nm and 270 nm in the UV region and a new prominent peak at 470 nm, denoting extension of conjugation. The blank reagent exhibits absorption maxima at 258 nm, 333 nm and 423 nm. The overlaid absorption spectra of nifedipine, RN, the azo adduct and CDNBD reagent are presented in Figure 1. The spectrum of the azo adduct shows a bathochromic shift with respect to both the nifedipine and RN due to chromophoric elongation caused by the new azo linkage. A hyperchromic shift observed in the spectrum of the azo adduct when compared with CDNBD is expected also due to a higher number of chromophores. The peak at 470 nm was selected as the analytical wavelength because at this wavelength, the difference in absorptivity between the blank reagent and the azo adduct was maximal.

Absorption spectra of nifedipine, RN, CDNBD and the azo adduct formed between RN and CDNBD.
Effect of diluting solvents
In studying the effect of diluting solvents using acetonitrile, ethanol, methanol and 1-propanol, the highest absorbance value was obtained with acetonitrile as the diluting solvent. This might be so because the alcohols are protic solvents promoting the formation of hydrogen bonding which invariably results in lower absorbance values due probably to reduction in the propensity of azo dye formation by interaction with the amino group on the residual nifedipine molecule to generate NH3+ which reduces the directing effects of the group for the in-coming CDNBD molecule. For acetonitrile, however, which is a polar aprotic solvent, its inability to promote hydrogen bonding formation probably results in the absorbance value being higher.
Optimisation studies
The first set of optimisation studies conducted was the determination of the optimum temperature and time required for the diazo coupling reaction between RN and CDNBD. As shown in Figure 2, there was an increase inabsorbance from 30°C to 60°C at both 5 and 20 min incubation period. Although there was no significant difference in the absorbance produced at 50°C and 60°C (i.e. 0.390 and 0.398 at 5 min; 0.395 and 0.4 at 20 min, respectively). Thus, 50°C was selected as the coupling reaction temperature. After 60°C, there was a decrease in absorbance. This reduction in absorbance at a temperature greater than 60°C could have been due to thermal decomposition of the azo adduct formed as previously noted in the utilisation of CDNBD as a derivatizing reagent. A temperature of 50°C was selected as being the optimum temperature for the reaction of RN with CDNBD. Optimisation of reaction time was thereafter carried out at 50°C. It was observed as shown in Figure 3 that the absorbance value peaked at 15 min, and thereafter, absorbance values reduced with increase in time up to a final decline at 30 min. Thermal decomposition of the azo adduct may also be responsible as observed for the assessment of optimal temperature. Thus, optimal conditions for the coupling reaction were established at 50°C and 15 min.

Optimization of coupling reaction temperature.

Optimization of coupling reaction time at 50°C.
Reaction stoichiometry
The optimal ratio for the formation of the azo adduct was investigated using Job's method of continuous variation. The result is presented in Figure 4. The highest possible absorbance reading was obtained at a mole fraction of 0.67 for the reagent solution, and the absorbance was found to decrease at lower or higher mole fractions. This shows that CDNBD and RN react in a 2:1 stoichiometric ratio. From our previous work, it has been discovered that higher diazonium concentration is necessary for azo dye formation [44]. It is therefore proposed that a single adduct is generated, but a higher reagent ratio is required for optimal detector response. Any of the reaction steps taking place in situ may have also required a higher mole fraction of the CDNBD reagent, leading subsequently to the 2:1 reagent to drug ratio.

Stoichiometric ratio determination for the formation of azo adduct between reduced nifedipine and CDNBD.
Validation of the new colorimetric method
For validation studies, calibration lines were prepared on three successive days, and the average absorbance readings were used to describe the linear regression line for the assay of nifedipine by the CDNBD method. The assays of nifedipine were linear over the range of 2.9 to 14.5 μg/mL of nifedipine with a linear regression of Y = 0.0214X + 0.0061 and a correlation coefficient of 0.9985. The 95% confidence intervals for the slope and intercept are in the range of 0.0214 ± 9.86 × 10−4 and 0.0061 ± 5.09 × 10−4, translating to 4.6% and 8.3%, respectively. The limits of detection (LOD) and quantitation (LOQ) were estimated as stipulated by the ICH guidelines [47] using the expressions below:
where σ is the standard deviation of the blank signals, and s is the slope of the calibration graph. The LOD and LOQ values obtained respectively are 0.1344 μg mL−1 and 0.4074 μg mL−1. The Sandell sensitivity obtained is 4.673 ng/cm2. The various analytical and validation parameters are summarised in Table 1. The values obtained for the LOD, LOQ and Sandell's sensitivity point to the highly sensitive nature of this new colorimetric method for the determination of nifedipine in bulk and tablet dosage forms. It is one of the recently reported methods for nifedipine with such a high sensitivity and makes it hold a promise for the quantitative determination of this important cardiovascular agent.
Accuracy and precision were estimated over 3 days by recovery studies. The intra- and inter-day data obtained are presented in Tables 2 and 3, respectively. Accuracy, as estimated by relative errors, ranged from 1.15% to 3.22% for intra-day assessment and −0.008% to 3.22% for inter-day assessment. The precision was generally good with a relative standard deviation of less than 3.1% for both intra- and inter-day assessment. The percentage recoveries for all analyte levels both at intra- and inter-day assessment were all close to 100%, justifying the suitability of this new colorimetric method.
Interference studies
The influence of commonly utilised excipients on the recovery of nifedipine from the reaction medium was studied in the presence of common excipients. The recoveries of nifedipine are as follows: starch, 103.98 ± 0.23%; lactose, 102.64 ± 0.27%; magnesium stearate, 101.30 ± 0.23%; gelatin, 103.98 ± 0.23; talc, 102.64 ± 0.27%; and a mixture of these excipients, 103.98 ± 0.23% (n = 6 in all determinations). With the good recoveries obtained, it implies that the method is free from undue interference by the commonly utilised tablet excipients.
Application to analysis of tablets
The new method was applied to the assay of nifedipine in six tablets brands. The results obtained as well as that of the official USP HPLC method are presented in Table 4. There was found to be no significant difference in the content of nifedipine in the tablets by both methods, thereby establishing the equivalence of both methods.
Relative merits of the new method
The new method described in this work has the advantage of being relatively simple compared to the official HPLC method, and it utilises a more readily available instrumentation which can be utilised in any environment for the quality control of nifedipine tablets. When compared to other previously reported spectrophotometric methods, this new colorimetric method has the advantage of circumventing extraction with organic solvents which may compromise accuracy. The method is also relatively simple as it adopts a two-step process, and it is highly sensitive compared to previous methods. This new colorimetric method is the first reported spectrophotometric method for the analysis of nifedipine based on azo dye formation.
Experimental
Chemicals and reagents
Acetonitrile, ethanol, methanol, 1-propanol, distilled water, glacial acetic acid, Zinc dust, concentrated HCl (all analar reagent grade obtained from BDH Chemical Ltd., Poole, England) and nifedipine reference standard were utilised in this study.
Nifedipine tablet brands assayed using this new colorimetric method include the following: Frapine Retard 20 mg/tablet (Medrel Pharmaceutical Ltd.), Nifedin Dexcel Retard 20 mg/tablet (Dexcel Ltd.), Nifegem Retard 20 mg/tablet (Gemini Pharmaceuticals Ltd.), Nifecard Retard 20 mg/tablet (Salutas Pharma GmbH), Medipine Retard 20 mg/tablet (Cipla Ltd.) and Nife-D 20 mg/tablet (Lincoln Pharmaceuticals Ltd.). All the brands were within their shelf-life periods during analyses.
Equipment
The following equipment were used: analytical balance (Mettler H80, Mettler-Toledo, Inc., Columbus, OH, USA), precoated TLC aluminium plate, ultraviolet lamp 254/364 nm (PW Allen and Co., London, UK), digital colorimeter (Model 6051, Jenway Ltd., Dunmow, Essex UK), UV-Visible spectrophotometer (PerkinElmer Lamda 25, Cambridge, UK), vortex mixer (Griffin and George Ltd., Loughborough, UK) and thermostated water bath (Langford, UK).
Statistical analysis
Statistical analysis was done between the results obtained from the new colorimetric method and the USP HPLC assay procedure using Origin version 6.1 (OriginLab Corporation, Northampton, MA, USA). P values less than or equal to 0.05 were taken as significant using the variance ratio (F test) and Student's t test.
Conclusions
The new colorimetric method is the first described method for the analysis of nifedipine by azo dye derivatization methodology. It has low concentrations of calibration ranges compared to the other existing photometric methods. There was no interference from common excipients encountered, and it is of equivalent accuracy with the official USP HPLC method. The method is simple, highly sensitive, accurate, precise, economical and suitable for routine analysis of nifedipine in bulk and drug formulations.
Authors’ information
SAA holds a Bachelor of Pharmacy degree and MSc in Pharmaceutical chemistry. He is at present a doctoral student in the Department of Pharmaceutical Chemistry, University of Ibadan, where he is a Lecturer.
OAA holds a PhD in Pharmaceutical analysis and Synthetic Chemistry. He is currently the Head of Department of Pharmaceutical Chemistry, University of Ibadan. He is a Senior Lecturer in the department. He researches actively in the area of derivatization methodology.
OSI is an Associate Professor in the Department of Pharmaceutical Chemistry. He holds a PhD degree in Pharmaceutical Chemistry from the same department. He researches actively in the field of Process Understanding and Modelling.
References
Sweetman S (Ed): Martindale: the complete drug reference. 36th edition. The Pharmaceutical Press, London; 2009:1350–1356.
Merck Research Lab, division of Merck and Co: The Merck Index. 12th edition. Whitehouse Station; 1996:1121.
British Pharmacopoeia Commission: British Pharmacopoeia, vol 2–3. The Pharmaceutical Press, London; 2010:1505–2939.
US Pharmacopoeial Convention Inc: United States Pharmacopoeia USP 30/NF 25. Rockville; 2000:2753.
Yuan G, Zhu B: Zhongguo Yiyao Gongye Zazhi. 1996, 27: 171.
Vyas SP, Goswami SK: Indian Drugs. 1993, 30: 342.
Mallick S, Gupta BK, Ghoshal SK: The Eastern Pharmacist. 1998, 129.
Umapathi P: Int J Pharm. 1994, 108: 11. 10.1016/0378-5173(94)90411-1
El-Walily AFM: J Pharm Biomed Anal. 1997, 16: 21. 10.1016/S0731-7085(96)01961-9
Kasture AV, Ramteke M: Ind J Pharm Sci. 2005, 67: 752.
Rahman N, Azmi SNH: Acta Pharm. 1999, 49: 113.
Hemmateenejada B, Miri R, Kamali R: J Iran Chem Soc. 2009, 6: 113. 10.1007/BF03246509
Karadi AB, Ravi KUM, Shobha M, Raju S: East Pharm. 2000, 117.
Rahman N, Hoda MN: Farmaco IL. 2002, 57: 435. 10.1016/S0014-827X(02)01228-4
Mahadik KR, Byale GB, More HN, Kadam SS: J Inst Chem. 1991, 63: 218.
Rahman N, Khan NA, Azmi SNH: Farmaco IL. 2004, 59: 47. 10.1016/j.farmac.2003.10.001
Rahman N, Najmul S, Azmi H: Acta Biochimica Polonica. 2005, 52: 915.
Sastry CSP, Chintalapati R, Venkateswarlu R: J Inst Chem. 1997, 69: 187.
Golcu AY, Serin S: Sci Pharm. 1998, 66: 341.
Revathi R, Ethiraj T, Saravanan VS, Ganeshan V, Saranya V, Sengottuvel T: International Journal of Pharmacy and Pharmaceutical Sciences 2. 2010.
Wang Z, Tang X, Hou J, Pan C, Wei X: Shenyang Yaoke Daxue Xuebao. 1981, 19: 38.
Rao GR, Raghuveer S, Srivastava CMR: Indian Drugs. 1985, 22: 435.
Beaulieu N, Curran NM, Graham SJ, Sears RW, Lovering EG: J Liq Chromatogr. 1991, 14: 1173. 10.1080/01483919108049311
Ohkubo T, Noro H, Sugawara K: J Pharm Biomed Anal. 1992, 10: 67. 10.1016/0731-7085(92)80012-C
Sha Z, Gao H, Sun W, Tian D: Zhongguo Yiyao Gongye Zazhi. 1993, 24: 176.
Barbato E, Cappelo B, Grumetto L, Morrica P: Farmaco IL. 1993, 48: 417.
Liu C, Chen G: Yaowu Fenxi Zachi. 1993, 13: 314.
Potter H, Hulm M: Pharm Biomed Anal. 1988, 61: 115.
Patravale VB, Nair VB, Gore SP: J Pharm Biomed Anal. 2000, 23: 623. 10.1016/S0731-7085(00)00317-4
Bretnall AE, Clarke GS: J Chromatogr A. 1995, 700: 173. 10.1016/0021-9673(95)00112-Z
Dumitrescu V, David V, Pavel A: Rev Chim. 2001, 52: 317.
Tompe P, Halbauer-Nagy A: Acta Pharm Hung. 1990, 60: 130.
Ghoneim MM, Tawfik A, Khashaba PY: Anal Bioanal Chem. 2003, 375: 369.
Richter P, Toral MI, Quiroz G, Jaque P: Lab Rob Autom. 1997, 9: 255. 10.1002/(SICI)1098-2728(1997)9:5<255::AID-LRA5>3.0.CO;2-#
Kumazawa T, Sato K, Seno H, Suzuki O: Hochudoku. 1993, 11: 128.
Idowu SO, Tambo SC, Adegoke AO, Olaniyi AA: Trop J Pharm Res. 2002, 1: 15.
Idowu OS, Adegoke OA, Olaniyi AA: J AOAC Int. 2004, 87: 573.
Idowu OS, Adegoke OA, Oderinu BA, Olaniyi AA: Pakistan J Pharm Sci. 2006,19(2006):134.
Adegoke OA, Idowu SO, Olaniyi AA: Acta Pharm. 2006, 56: 189.
Adegoke OA, Idowu OS, Olaniyi AA: J Iran Chem Soc. 2006, 3: 277. 10.1007/BF03247220
Adegoke OA, Idowu SO, Olaniyi AA: Trop J Pharm Res. 2007, 6: 695.
Adegoke AO, Idowu SO, Olaniyi AA: Afr J Med Sci. 2007, 36: 249.
Idowu OS, Adegoke OA, Adeniji AO, Olaniyi AA: East Cent Afri J Pharm Sc. 2009, 12: 8.
Adegoke OA, Idowu OS, Daramola OP, Ogunsanya OS: Acta Pharm Scien. 2010, 52: 269.
Idowu OS, Kolawole AO, Adegoke OA, Kolade YT, Fasanmade AA, Olaniyi AA: J AOAC Int. 2005, 88: 1108–1113.
Smith MB, March J: March's advanced organic chemistry: reactions, mechanisms, and structure. 16th edition. Wiley, Inc, Hoboken; 2007:1709.
European Medicines Agency:ICH Topic Q2 (R1) Validation of analytical procedures: text and methodology (CPMP/ICH/281/95). 1995. [http://www.emea.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002662.pdf.Accessed10Jan2011]
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SAA carried out the experimental studies and prepared the first draft of the manuscript. OAA initiated the study, prepared the design of the study and supervised the acquisition of the data carried out the data analysis, prepared the graphics and prepared the manuscript for submission, review and final acceptance. OSI provided general logistics support. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Aderibigbe, S.A., Adegoke, O.A. & Idowu, O.S. A new colorimetric method for the determination of nifedipine tablets by derivatization using 4-carboxyl-2,6-dinitrobenzene diazonium ion. Int J Ind Chem 3, 5 (2012). https://doi.org/10.1186/2228-5547-3-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2228-5547-3-5