
Abstract
Tumor necrosis factor alpha (TNF-α) was discovered more than a century ago, and its known roles have extended from within the immune system to include a neuro-inflammatory domain in the nervous system. Neuropathic pain is a recognized type of pathological pain where nociceptive responses persist beyond the resolution of damage to the nerve or its surrounding tissue. Very often, neuropathic pain is disproportionately enhanced in intensity (hyperalgesia) or altered in modality (hyperpathia or allodynia) in relation to the stimuli. At time of this writing, there is as yet no common consensus about the etiology of neuropathic pain - possible mechanisms can be categorized into peripheral sensitization and central sensitization of the nervous system in response to the nociceptive stimuli. Animal models of neuropathic pain based on various types of nerve injuries (peripheral versus spinal nerve, ligation versus chronic constrictive injury) have persistently implicated a pivotal role for TNF-α at both peripheral and central levels of sensitization. Despite a lack of success in clinical trials of anti-TNF-α therapy in alleviating the sciatic type of neuropathic pain, the intricate link of TNF-α with other neuro-inflammatory signaling systems (e.g., chemokines and p38 MAPK) has indeed inspired a systems approach perspective for future drug development in treating neuropathic pain.
Similar content being viewed by others

Introduction
Despite intense research over the last 30 years, debate is still ongoing regarding the nature of neuropathic pain, including controversy as to whether such pain is peripheral or central in origin, and as to whether its etiology is inflammatory or non-inflammatory. Increasing evidence has provided better understanding of the roles of both immune and pro-inflammatory mediators (e.g., the interleukins, TNF-α, complement components, ATP and the chemokines) in the mechanisms of both peripheral and central neuropathic pain [1–4]. This review will concentrate on current knowledge and experimental models regarding the role of TNF-α, among other cytokines, in neuropathic pain; with an appraisal of available potential therapeutic targets related to TNF-α and directions for future developments in this area.
Neuropathic pain as an example of an inflammatory pain model
Neuropathic pain is characterized by disproportionate hypersensitivity to stimuli (hyperalgesia), abnormal pins-and-needles or electric-shock-like sensations (hyperpathia) and, finally, nociceptive responses to non-noxious stimuli (allodynia). It is a pathological type of pain that persists despite resolution of the inciting damage to the nerve and the surrounding tissues. From a behavioral standpoint, nociception is an adaptive tool for better survival, while neuropathic pain is considered maladaptive. The prevalence of neuropathic pain ranges from 1% in UK [5] to 1.5% in the US [6] to 17.9% in Canada [7]. Weir Mitchell [8] is often credited with the first descriptive account of neuropathic pain from nerve injuries seen in the US Civil War, using terms that range from "burning", "mustard red hot", "red-hot file rasping the skin" to "with intensity ranging from most trivial burning to a state of torture". Clinically, the top three most common types of neuropathic pain are post-herpetic neuralgia, trigeminal neuralgia and diabetic neuropathy [9]. Neuropathic pain is among the most difficult types of chronic pain to treat, which not only significantly impairs patients' quality of life [10] but also adds to the burden of direct and indirect medical cost for our society [10, 11]. Conceptually, neuropathic pain consequent to peripheral nerve injury results from an increased excitability of the neurons as a result of sensitization. The debate is still on-going as to whether this sensitization occurs in the peripheral or central compartments of the nervous system, or both. Experimentally, various animal models of peripheral neuropathic pain have been developed: chronic constriction injury (CCI) of the sciatic nerve with loose ligatures [12–15]; partial sciatic nerve injury with tight ligatures [15–17]; total sciatic nerve ligation [15, 18]; sciatic nerve transaction [19–21] and axotomy of lumbar roots entering the sciatic nerve [22, 23]. Despite the various degrees and modes of nerve damage in these models, there is a common sequel--post-injury inflammatory changes leading to mast cell degranulation [24], and recruitment of both macrophages [25] and polymorphonuclear neutrophils [26]. However, in CCI models thermal hyperalgesia still occurs when ligatures are loosely placed around the sciatic nerve without actual mechanical damage [27]. This finding supports the hypothesis that it is the inflammatory microenvironment [28] and the release of mediators [29], rather than the nerve injury per se, that is pivotal for the development of neuropathic pain. Clatworthy et al. [30] further demonstrated that suppression of the inflammatory response with dexamethasone reduces thermal hyperalgesia, while enhancing the inflammatory response using Freud's adjuvant was seen to aggravate the level of pain hypersensitivity. His work set the stage for continuing research on immune and pro-inflammatory mediators in neuropathic pain over the next two decades. An updated list of such mediators, by no means exhaustive, includes the eicosanoids [31–34], bradykinins [35, 36], serotonin [37–39], ATP/ADP [40–42], neurotrophins [43–46], cytokines [47–52], chemokines [53, 54], and reactive oxygen species [21, 55, 56]. These mediators are not exclusive to cells of immune/inflammatory origin, but are also produced by Schwann cells [57–59] and spinal glial cells [42, 60–63], thereby potentially mediating the mechanisms of neuropathic pain.
Cytokines in neuropathic pain
Cytokines are low molecular weight glycoproteins that are secreted mainly, but not exclusively, by immunological cells such as T-cells, macrophages and neutrophils. Other cells that secrete cytokines include keratinocytes and dendritic cells of the skin [64] and Schwann cells and glial cells of the central nervous system [65, 66]. They act as intercellular mediators regulating the functions and differentiation of neighboring cells and are produced in response to disease, inflammation, or tissue damage. Cytokine synthesis is prompt and their actions are often localized with a relatively short half-life. This distinguishes them from hormones which are constantly produced with longer-lasting and more distant effects. The first cytokine was discovered by Beeson in 1948 [67] as a pyrogenic compound extracted from ploymorphonuclear leucocytes, later known as IL-1β. Since then, many other cytokines have been discovered, and these fall into five main categories: interleukins, interferons, tumor necrosis factors, growth factors and chemokines. Together, these factors contribute to the pathogenesis of neuropathic pain [47, 68]. In particular, tumor necrosis factor alpha (TNF-α) [69, 70], interleukin-1 (IL-1) [47, 71, 72] and interleukin-6 (IL-6) [49, 73] have been associated with the development of neuropathic pain in various animal models [74]. In this review, we shall limit our scope to TNF-α.
Tumor necrosis factor alpha (TNF-α): a neuropathic pain-related cytokine
In 1891, the success story of William Coley in using supernatant extract of heat-killed mixtures of Streptococcus pyogenes and Serratia marcescens bacteria to treat tumors may in fact be the first discovery of tumor necrosis factor [75]. It was not until 1975 that an endotoxin-like substance was described in activated macrophages with tumor-regression activity and was given the name of tumor necrosis factor alpha, TNF-α [76]. TNF-α belongs to a superfamily of ligand/receptor proteins called the tumor necrosis factor/tumor necrosis factor receptor superfamily proteins (TNF/TNFR SFP). TNF-α possess a trimeric symmetry with a structural motif called the TNF homology domain (THD), which is shared with all other members of the TNF proteins. This THD binds to the cysteine-rich domains (CRDs) of the TNF receptors (TNFRs), and variations of these CRDs lead to heterogeneity of the TNFRs [77]. TNFRs are either constitutively expressed (TNFR1, p55-R) or inducible (TNFR2, p75-R) [78]. In the context of neuropathic pain, using the standard model of chronic constriction injury (CCI) of sciatic nerve in rats, TNF-α has been detected at the injury site and shows temporal up-regulation [79–81]; here TNF-α is located mainly in macrophages [82] and Schwann cells [70, 83] by immuno-reactive staining. Similarly, there is local up-regulation of both TNFR1 and TNFR2 as injured neurons undergo Wallerian degeneration, albeit at differential rates [84]. Similar results are found in humans, where nerve biopsies from patients with painful neuropathy show higher levels of TNF-α expression, especially in Schwann cells [85]. Intra-sciatic injection of TNF-α in rats reproduces pain hypersensitivity that is similar to that of neuropathic pain in humans [69, 86], and this is reversible with neutralizing antibodies to TNFR [86], in particular TNFR1 [50]. TNF-α enhances the tetrodotoxin-resistant (TTX-R) Na+ current in cultured DRG cells from wild-type but not from TNFR1-knockout mice, and such current is abolished by a p38-MAPK inhibitor; implying that TNF acts via TNFR1 and activates TTX-R Na+ channels via the p38 MAPK system [87]. Further studies using TNFR1/TNFR2 knock-out mice have suggested a neurotoxic role for TNFR1 versus a neuroprotective role of TNFR2 [88]. However, there is still debate regarding the relative roles of TNFR1 and TNFR2 in chronic pain: in mice with tumor-induced thermal hyperalgesia, deletion of the TNFR2 gene reduces the painful response hence signifying a role for TNFR2 [89]; whilst in rats with spinal root injury, TNFR1 elicits excitatory responses in DRG of adjacent uninjured roots and TNFR2 excites DRG neurons from injured roots [90]. In the inflammatory models of carrageenan-induced and zymosan-induced pleurisy in rat models, TNF-α has been found to have a lead role in activating a cascade of other cytokines, notably IL-1β, IL-6 and IL-8 [91]. A similar local cascade has been demonstrated in a model of neuropathic pain following nerve injury [83].
The role of TNF-α in peripheral mechanisms of neuropathic pain
TNF-α plays a role in the peripheral mediation of neuropathic pain. Clinically, HIV therapy and chemotherapy produce peripheral neuropathy with massive release of TNF-α in serum [92] and TNF-α used as a clinical anti-cancer treatment leads to peripheral neuropathy [93]. Traditional CCI of sciatic nerve in rats results in raised levels of TNF immunoreactivity in dorsal root ganglia (DRG) of both injured and uninjured ipsilateral adjacent afferents [94], as well as of contralateral uninjured counterparts [95], which can only be partly explained by retrograde axonal transport [96]. There is also a corresponding up-regulation of TNFR1 and TNFR2 in both nerve and DRG [97], with a temporal pattern of increased TNF mRNA expression, first in sciatic nerve, and then in DRG [98]. When nucleus pulposus extract of coccxygeal intervertebral disc is applied to lumbar DRG of rats, neuropathic pain is induced but is abolished by co-application of TNRF1, implying a direct role of TNF as a local mediator [99]. Exogenous TNF-α injected into DRG of CCI roots is transported both anterograde to the site of injury and retrograde into the dorsal horn [100], precipitating allodynia in both the ligated and adjacent uninjured nerves [101]. TNF-α is known to lead to apoptosis via TNFR1 [102, 103] and the caspase signaling pathway [103]. Caspase inhibitors can attenuate peripheral neuropathy experimentally induced by HIV therapy or chemotherapy in rats [104]. A recent study compared crush injury of L5 spinal nerve (distal to DRG) with L5 nerve roots (proximal to DRG) in rats and found that distal crush injury resulted in more neuronal apoptosis and enhanced TNF-α expression and caspase levels, correlating with higher neuropathic pain [105], lending more support to a TNF-α-apoptosis-caspase signaling paradigm for peripheral neuropathic pain. In addition to enhancing TTX-R Na+ channels in nociceptive DRG neurons [87], TNF-α can also increase membrane K+ ion conductance in a non-voltage-gated fashion [106] leading to overall neuronal hyper-excitability and hence leading to neuropathic pain.
The role of TNF-α and glia in central mechanisms of neuropathic pain
In late 1990s, TNF-α was proposed to be one pro-inflammatory cytokine with a pivotal role in the "immune-to-brain" pathway of communication for pain, and in models of sickness response in general [51, 107]. In classic CCI models in rats, increased levels of TNF-α are found in hippocampus [108, 109], locus coeruleus [109, 110] and red nucleus [111] of brain. Recent data have suggested that TNF-α mediates central mechanisms of neuropathic pain through glial systems. In the central nervous system, glial cells outnumber neurons by as much as 50-fold, and include three relevant types: astrocytes, oligodendrocytes and microglia. Oligodendrocytes not only provide the myelin sheaths that insulate the neurons, they also contribute to the actual expansion of neuronal caliber and reorganization of neurofilaments [112]. Astrocytes are the most abundant glial cells and possess the most diverse functions: they can modulate synaptic functions by forming a tripartite synapse with pre-synaptic, post-synaptic and extra-synaptic astrocytic contacts with up to 10,000 other neurons [113, 114] using glutamate and adenosine as neurotransmitters [115]. It has been suggested that spinal astrocytes may play a role in sensitization of chronic pain via activation of the p38-MAPK system [116, 117], and may even synapse with microglia, with pre-synaptic neuronal processes and with post-synaptic neuronal structures to form a tetrapartite configuration [118]. Astrocytes also regulate maturation of neurons and synpatogenesis, hence playing a pivotal role in modulation of neural plasticity [119]. Microglia constitute 15-20% of the total glial population and serve as an immune invigilator for the central nervous system. They originate from tmesodermal precursor cells of hemopoietic lineage. In response to nerve injury and inflammation, microglia transform into macrophage-like cells [120] that express major histocompatibility complex antigens and secrete pro-inflammatory cytokines, including TNF-α, IL-1 and IL-6 [121, 122]; CCL2 and CX3CL1 [53, 123], and ATP, which mediate their effects via the p38-mitogen-activated protein kinase (p38-MAPK) system [41, 124, 125].
Back in 1991, it was shown that classic CCI leads to hypertrophy of astroglia in the dorsal horn of spinal cord as reflected by increased immunostaining of glial fibrillary acidic protein [126]. Since then, other subcutaneous and intraperitoneal inflammatory pain models [127] have also been shown to induce glial activation. In newborn rats, where microglia are immature, intrathecal lipopolysaccharide (LPS) fails to evoke the allodynia response that is invariably seen in adult rats [62], suggesting a necessary role for functional microglia in the pathogenesis of neuropathic pain. Along with various other mediators, TNF-α has been shown to be present on the surfaces of astrocytes by immunofluorescence staining, where TNF-α auto-stimulates its own production via G-protein coupled receptor (CXCR4) and TNF-α converting enzyme. The result is a cascade of events leading typically to production of IL-1, IL-6, nitric oxide and ATP [121, 128], all of which contribute to enhanced neuronal activity leading to pathological pain. Wei et al [129] demonstrated increased levels of TNF-α and IL-1β in the rostral ventromedial medulla (RVM) of rats after CCI of the infraorbital nerve, with a corresponding enhancement of phosphorylation of the NR1 subunit of NMDA receptors, which is thought to be coupled to the receptors for both TNF-α and IL-1β. Injection of TNF-α and IL-1β into RVM increases NR1 phosphorylation of NMDA receptors and produces hyperalgesia, which is reversed by an NMDA antagonist. Wei's work sparked off research into NMDA receptors as a possible target for treating neuropathic pain; unfortunately, progress has been discouraged by the ubiquitous expression of NMDA receptors in the human central nervous system, which renders NMDA receptor blockade for analgesia an impossible task without concomitant alterations in cognition, memory and learning.
TNF-α, neural plasticity and neuropathic pain
Originally identified in hippocampus as a substrate for memory storage and learning, the synaptic mechanisms of long term potentiation (LTP) in glutamergic neurons [130] have since been demonstrated as well in other parts of the central nervous system; in particular, in the dorsal horn of the spinal cord, where they may lead to abnormal nociception and neuropathic pain [131, 132]. Normal nociceptive signals are conveyed by both Aδ and C-fibers; of which the latter make synapses with second-order neurons in the spinal dorsal horn. The LTP phenomenon has been well characterised in C-fibers of rat dorsal horn with tetanic stimulation [133, 134] and also with acute nerve injury [135]. High-frequency stimulation leads to an LTP pattern of cutaneous allodynia and hyperalgesia in humans [136] with a typical early LTP time course [137]. As the signalling mechanism of LTP unfolds, TNF-α is found to play an important role. Endogenous glial TNF-α can modulate synaptic plasticity by increasing the expression of AMPA receptors in cultured rat hippocampal slices [138] for homeostatic regulation of synaptic strength in an activity-dependent fashion [139]. However, TNF-α given at non-physiological levels often inhibits LTP in similar models of cultured rat hippocampus [140, 141]. As regards to C-fibers in the spinal dorsal horn, exogenous TNF-α produces LTP in C-fiber evoked field potentials only in the presence of nerve injury, and this LTP is blocked by inhibitors of NF-kappa B, JNK and p38-MAPK [142]. In the absence of nerve injury, TNF-α can neutralise the action of src-family kinase inhibitors by restoring LTP in C-fiber evoked potentials as normally induced by high-frequency stimulation (HFS).
TNF-α, ATP and p38-MAPK
Since the 1950s, release of ATP has been detected from nerve endings [143, 144] and a role for ATP in nociception was implicated when it was shown to induce pain in human blister bases [145]. ATP excites cutaneous afferent neurons of animal models in a fashion similar to that of other neurotransmitters like 5-HT and acetylcholine [146], and can act proximally to excite DRG neurons [147]. Around the same time, Burnstock and his colleagues [148, 149] first characterized purinergic receptors into P1 (sensitive to adenosine, ADO), P2X, and P2Y receptors (sensitive to ATP and ADP). Molecular cloning studies have identified four sub-types of P1 (A1, A2A, A2B, and A3), seven sub-types of P2X (P2X1 to P2X7) and 8 sub-types of P2Y receptors (P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13, P2Y14) [150]. Each subtype has a different distribution in neuronal and glial cells, interacting with each other in an intricate manner. In terms of signaling functions, P1 and P2Y receptors are G-protein coupled receptors, while P2X receptors are ligand-gated ion channels [151]. Within the context of neuropathic pain, P2X3, P2X4 and P2X7 receptors are thought to play a role; and in particular, P2X3 may act via the TTX-R voltage-gated sodium channel Nav 1.9 [152].
Earlier studies using nerve injury models in rats revealed either an increase [153] or decrease [154] of P2X3 immuno-reactivity of the DRG neurons, depending on the type of nerve injury. When expression of P2X3 receptors in DRG is reduced using anti-sense oligonucleotides [155] or siRNA [156], development of mechanical hyperalgesia is mitigated after classic CCI. Furthermore, administration of anti-sense oligonucleotides to knock down P2X3 receptors can reverse established neuropathic pain that re-emerges after cessation of the anti-sense treatment [157], suggesting a dynamic modulatory role of P2X3 receptors. Following a similar approach, Tsuda et al [158] demonstrated an increase in P2X4 receptor expression after chronic nerve injury, and showed that both pharmacological blockade and anti-sense oligonucleotide treatment abrogates the development of mechanical allodynia. Later studies have suggested that P2X4 receptor stimulation leads to secretion of brain-derived neurotrophic factor (BDNF) in spinal microglia, and that this BDNF is involved in mediating neuropathic pain [40, 159, 160], possibly via activation of the p38-MAPK system [161]. P2X7 receptors are associated with TNF-α production in microglia through the p38-MAPK system [162, 163], as an inhibitor of MAPK system will suppress production of TNF-α mRNA and an inhibitor of p38 will prevent nucleocytoplamic transport of TNF-α mRNA [162]. Independent of ATP, the p38-MAPK system seems to be essential for the action of TNF-α via TTX-R Na+ channels [87]. As an entity itself, microglial p38-MAPK has been implicated in the pathogenesis of neuropathic pain in studies using various in vivo models of peripheral nerve [164, 165] and spinal cord injury [166, 167]. For example, spinal nerve ligation in rats leads to allodynia with concomitant rises in TNF-α and p38 phosphorylation; treatment with inhibitors of either TNF-α or p38 results in reduction of allodynia and, finally, TNF-α blockade can in turn suppress p38 activation [168]. Studies using HSV-mediated gene transfer in nerve injury animal models have shown induced expression of soluble p55 TNFR (sTNFR2) in DRG neurons, resulting in decreased phosphorylation of p38 and reduced allodynia, again suggesting a causal link between TNF-α and the p38-MAPK system.
TNF-α as potential drug target for chronic pain--the possibilities
Due to the unique trimeric structure shared between the TNF ligand and the TNF receptor (both belonging to the TNF/TNFR SPF), the transmembrane portion of TNF molecule (mTNF), besides being a ligand, is capable of acting as a receptor for a soluble form of TNF (sTNF) in a "reverse-signaling" manner [169], which then inhibits phosphorylation of p38 and hence expression of TNF protein. This unique phenomenon makes it possible to use gene therapy with a herpes simplex virus vector carrying a p55 sTNFR gene to transfect DRG neurons of rats [170]. As a result, over-expressed p55 sTNFR (sTNFR2) binds to the mTNF of DRG and down-regulates overall production of TNF by reverse signaling, significantly reducing the allodynia and hyperalgesia responses to CCI [170, 171]. Following a similar logic, a fusion protein (ELP-sTNFR2) has been developed wherein a soluble form of TNFR2 (sTNFR2) is conjugated to a temperature-sensitive elastin-like polypeptide (ELP), which can be thermally triggered to form a deposit around the peri-neural site of injection [172]. This fusion protein has been reported to be able to mitigate levels of TNF-α in DRG of injured nerve in rat models [173]. Indeed, many studies have demonstrated that local or spinal administration of agents that antagonize TNF-α will attenuate pain behaviors in neuropathic animal models [174–177]. Mechanical allodynia in the rat model of central neuropathic pain due to T13 spinal cord hemisection is attenuated by immediate, but not delayed, intrathecal administration of etanercept (a fusion protein blocker of TNF-α) at 1-4 weeks post spinal cord injury [178]. Propentofylline is a methylxanthine that inhibits lipopolysaccharide (LPS)-induced release of both TNF-α and IL-1β in a dose-dependent manner in glial cultures [179] and abates allodynia in rat spinal nerve transection models by modulating glial activation [180, 181]. Propentofylline was initially evaluated for treating dementia [182], but was eventually withdrawn from further clinical studies due to patent issues [183], and its efficacy in animal neuropathic pain models has yet to be tested in humans. Thalidomide, once banned in 1963 due to its teratogenicity, is now regaining favor in neuropathic pain research due to its ability to cross the BBB and its inhibitory effects on TNF-α (in vitro and in-vivo) and on IL-1/IL-6 (in-vitro only) [184, 185]. In the rat model of CCI, systemic thalidomide reduces the hyperalgesia response coincident with reductions in TNF-α levels, unchanged levels of IL-1/IL-6 and increased levels of IL-10 [186, 187]. Clinically, there have been sporadic reports of success in using thalidomide to treat complex regional pain syndromes [188]. However, the balance of thalidomide's efficacy versus safety in treating in chronic and neuropathic pain needs further clinical study [189], especially in view of its paradoxical neurotoxicity [189, 190]. Methotrexate is a well-known drug for treating cancer that is derived from glutamic acid. It is capable of crossing the BBB [191] and has anti-rheumatoid and anti-inflammatory actions through its inhibition of production of TNF-α via adenosine nucleotides [192, 193] and its ability to antagonize the actions of IL-1 [194]. Intrathecal administration of methotrexate reduces classic CCI-induced allodynia in rats [195] but its value in treating neuropathic pain is severely offset by its propensity per se to induce astrocytic proliferation [196] and hence neurotoxicity [197, 198].
The role of TNF-α in chronic pain seems irrefutable in view of abundant data from various neuropathic animal models, and with the actual isolation of TNF-α from neuropathic nerves [85] and perineural fat from radiculopathic nerve roots [199] in humans. An initial pilot study using subcutaneous etanercept to treat patients admitted to the hospital with acute severe sciatica showed improved pain scores [200]. Similarly, an open-label study with infliximab (an antibody to TNF-α) revealed promising results [201]. Subsequent randomized controlled trials failed to support the benefits of systemic anti-TNF-α treatment [202–205], but a recent report did show positive benefits of epidurally administered etanercept in the treatment of sciatica [206]. To date we are unaware of any randomized controlled clinical trials of infliximab or etanercept in treating other types of neuropathic pain. AV411(ibudilast), a trial drug that was originally developed as a non-selective phosphodiesterase inhibitor for treating bronchial asthma, has been studied in phase I and phase 2a clinical trials in the US and in Australia for treatment of diabetic neuropathic pain [207], based on findings that AV411 also suppresses glial cell activation and reduces the production of pro-inflammatory cytokines (IL-1β, TNF-α, IL-6) in rat neuropathic pain model [208].
Perspective on future studies
TNF-α is undoubtedly a titan in the research of neuropathic pain, and is by no means the only one in the arena. It is a pivotal member of the cytokine mediator system that is intrinsic to the pathogenesis of neuropathic pain both at peripheral and central levels (See Fig 1). Together with other mediators like interleukins, nerve growth factors, chemokines and interferons, it forms a network that interacts with downstream signaling mechanisms like the NMDA, ATP and MAPK systems. We now know that removing TNF-α from the picture will not abolish neuropathic pain as has already been demonstrated by the failure of TNF-α antagonists in clinical trials for sciatica [202–205]. Emerging data have guided research towards a collective role for glia-derived mediators and their coupled signaling pathways in the modulation of neuropathic pain [122, 127, 209, 210]. The paradigm is shifting from a single compound towards a system as a potential target for novel drug development for treating neuropathic pain [211–214]. Examples include the chemokine system [215], the MAPK system[216], and the glial system as a whole [217].

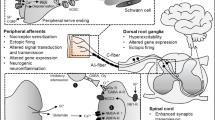
The roles of TNF-α as recognized at different levels of the nervous system in neuropathic pain induced by nerve injury: (1) at site of nerve injury; (2) at dorsal root ganglion; (3) at dorsal horn of the spinal cord; and (4) at the brain and higher centres.
Abbreviations
- 5-HT:
-
5-hydroxytryptamine (Serotonin)
- ADO:
-
Adenosine
- ADP:
-
Adenosine diphosphate
- ATP:
-
Adenosine triphosphate
- BBB:
-
Blood-brain barrier
- BDNF:
-
Brain-derived neurotrophic factor
- CCI:
-
Chronic constrictive injury
- CCL2:
-
Chemokine (C-C motif) ligand-2
- CX3CL1:
-
Chemokine (C-X3-C motif) ligand 1
- CXCR4:
-
CXC chemokine receptor-4
- DRG:
-
Dorsal root ganglion
- CRDs:
-
Cysteine rich domains
- ELP:
-
Elastin-like polypeptide
- ERK:
-
Extracellular signal-regulated kinases
- HFS:
-
High Frequency stimulation
- HSV:
-
Herpes simplex virus
- IL-1:
-
Interleukin-1
- IL-1β:
-
Interleukin-1 beta
- IL-6:
-
Interleukin-6
- IL-8:
-
Interleukin-8
- IL-10:
-
Interleukin-10
- JNK:
-
c-Jun N-terminal kinases
- LPS:
-
Lipopolysaccaride
- LTP:
-
Long Term Potentiation
- MAPK:
-
Mitogen activated protein kinase
- NMDA:
-
N-methyl-D-aspartic acid
- NSAIDs:
-
Non-steroidal anti-inflammatory drugs
- siRNA:
-
Small interfering RNA
- RVM:
-
Rostral ventromedial medulla
- THD:
-
TNF homology domain
- mTNF:
-
Transmembrane portion of TNF
- sTNF:
-
Soluble form of TNF
- TNF-α:
-
Tumor necrosis factor alpha
- TNFR:
-
Tumor necrosis factor receptor
- sTNFR2:
-
Soluble p55 TNF receptor
- TNFR SFR:
-
Tumor necrosis factor receptor super family receptor
- TTX-R:
-
Tetrodotoxin resistant.
References
Campbell JN, Meyer RA: Mechanisms of neuropathic pain. Neuron. 2006, 52: 77-92. 10.1016/j.neuron.2006.09.021.
McMahon SB, Cafferty WB, Marchand F: Immune and glial cell factors as pain mediators and modulators. Exp Neurol. 2005, 192: 444-462. 10.1016/j.expneurol.2004.11.001.
Marchand F, Perretti M, McMahon SB: Role of the immune system in chronic pain. Nat Rev Neurosci. 2005, 6: 521-532. 10.1038/nrn1700.
Moalem G, Tracey DJ: Immune and inflammatory mechanisms in neuropathic pain. Brain Res Rev. 2006, 51: 240-264. 10.1016/j.brainresrev.2005.11.004.
Bowsher D: Neurogenic pain syndromes and their management. Br Med Bull. 1991, 47: 644-666.
Cartier GT, Galer BS: Advances in the management of neuropathic pain. [Phys Med Rehabil Clin N Am. 2001] - PubMed result. Phys Med Rehabil Clin N Am. 2001, 12: 447-459.
Toth C, Lander J, Wiebe S: The prevalence and impact of chronic pain with neuropathic pain symptoms in the general population. Pain Med. 2009, 10: 918-929. 10.1111/j.1526-4637.2009.00655.x.
Mitchell SW: Injuries of nerves and their consequences. Philadelphia, Lippincott. 1872
Hall GC, Carroll D, Parry D, McQuay HJ: Epidemiology and treatment of neuropathic pain: the UK primary care perspective. Pain. 2006, 122: 156-162. 10.1016/j.pain.2006.01.030.
McDermott AM, Toelle TR, Rowbotham DJ, Schaefer CP, Dukes EM: The burden of neuropathic pain: results from a cross-sectional survey. Eur J Pain. 2006, 10: 127-135. 10.1016/j.ejpain.2005.01.014.
O'Connor AB: Neuropathic pain: quality-of-life impact, costs and cost effectiveness of therapy. Pharmacoeconomics. 2009, 27: 95-112. 10.2165/00019053-200927020-00002.
Bennett GJ, Xie YK: A peripheral mononeuropathy in rat that produces d... [Pain. 1988] - PubMed result. Pain. 1988, 33: 87-107. 10.1016/0304-3959(88)90209-6.
Xie Y, Zhang J, Petersen M, LaMotte RH: Functional changes in dorsal root ganglion cells after chronic nerve constriction in the rat. J Neurophysiol. 1995, 73: 1811-1820.
Kingery WS, Castellote JM, Wang EE: A loose ligature-induced mononeuropathy produces h... [Pain. 1993] - PubMed result. Pain. 1993, 55: 297-304. 10.1016/0304-3959(93)90004-9.
Kim KJ, Yoon YW, Chung JM: Comparison of three rodent neuropathic pain models. Exp Brain Res. 1997, 113: 200-206. 10.1007/BF02450318.
Shir Y, Seltzer Z: A-fibers mediate mechanical hyperesthesia and allo... [Neurosci Lett. 1990] - PubMed result. Neurosci Lett. 1990, 115: 62-67. 10.1016/0304-3940(90)90518-E.
Seltzer Z, Dubner R, Shir Y: A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain. 1990, 43: 205-218. 10.1016/0304-3959(90)91074-S.
Nuytten D, Kupers R, Lammens M, Dom R, et al: Further evidence for myelinated as well as unmyeli... [Exp Brain Res. 1992] - PubMed result. Exp Brain Res. 1992, 91: 73-78. 10.1007/BF00230014.
Puke MJ, Xu XJ, Wiesenfeld-Hallin Z: Intrathecal administration of clonidine suppresses... [Neurosci Lett. 1991] - PubMed result. Neurosci Lett. 1991, 133: 199-202. 10.1016/0304-3940(91)90569-F.
Miao P, Madec K, Gong Y, Shen H, Eisenstat D, Melanson M, Gu X, Leong C, Klowak M, Namaka M: Axotomy-induced up-regulation of tumor necrosis factor-alpha in the dorsal root ganglia. Neurol Res. 2008, 30: 623-631. 10.1179/174313208X289606.
Guedes RP, Araujo AS, Janner D, Bello-Klein A, Ribeiro MF, Partata WA: Increase in reactive oxygen species and activation of Akt signaling pathway in neuropathic pain. Cell Mol Neurobiol. 2008, 28: 1049-1056. 10.1007/s10571-008-9279-9.
Li L, Xian CJ, Zhong JH, Zhou XF: Effect of lumbar 5 ventral root transection on pain behaviors: a novel rat model for neuropathic pain without axotomy of primary sensory neurons. Exp Neurol. 2002, 175: 23-34. 10.1006/exnr.2002.7897.
Xu JT, Xin WJ, Zang Y, Wu CY, Liu XG: The role of tumor necrosis factor-alpha in the neuropathic pain induced by Lumbar 5 ventral root transection in rat. Pain. 2006, 123: 306-321. 10.1016/j.pain.2006.03.011.
Olsson Y: Degranulation of mast cells in peripheral nerve injuries. Acta Neurol Scand. 1967, 43: 365-374. 10.1111/j.1600-0404.1967.tb05739.x.
Perry VH, Brown MC, Gordon S: The macrophage response to central and peripheral nerve injury. A possible role for macrophages in regeneration. J Exp Med. 1987, 165: 1218-1223. 10.1084/jem.165.4.1218.
Daemen MA, Kurvers HA, Kitslaar PJ, Slaaf DW, Bullens PH, Wildenberg Van den FA: Neurogenic inflammation in an animal model of neuropathic pain. Neurol Res. 1998, 20: 41-45.
Maves TJ, Pechman PS, Gebhart GF, Meller ST: Possible chemical contribution from chromic gut sutures produces disorders of pain sensation like those seen in man. Pain. 1993, 54: 57-69. 10.1016/0304-3959(93)90100-4.
Sommer C, Galbraith JA, Heckman HM, Myers RR: Pathology of experimental compression neuropathy producing hyperesthesia. J Neuropathol Exp Neurol. 1993, 52: 223-233. 10.1097/00005072-199305000-00006.
Frisen J, Risling M, Fried K: Distribution and axonal relations of macrophages in a neuroma. Neuroscience. 1993, 55: 1003-1013. 10.1016/0306-4522(93)90314-6.
Clatworthy AL, Illich PA, Castro GA, Walters ET: Role of peri-axonal inflammation in the development of thermal hyperalgesia and guarding behavior in a rat model of neuropathic pain. Neurosci Lett. 1995, 184: 5-8. 10.1016/0304-3940(94)11154-B.
Schafers M, Marziniak M, Sorkin LS, Yaksh TL, Sommer C: Cyclooxygenase inhibition in nerve-injury- and TNF-induced hyperalgesia in the rat. Exp Neurol. 2004, 185: 160-168. 10.1016/j.expneurol.2003.09.015.
Ma W, Du W, Eisenach JC: Role for both spinal cord COX-1 and COX-2 in maintenance of mechanical hypersensitivity following peripheral nerve injury. Brain Res. 2002, 937: 94-99. 10.1016/S0006-8993(02)02593-3.
Syriatowicz JP, Hu D, Walker JS, Tracey DJ: Hyperalgesia due to nerve injury: role of prostaglandins. Neuroscience. 1999, 94: 587-594. 10.1016/S0306-4522(99)00365-6.
Okubo M, Yamanaka H, Kobayashi K, Noguchi K: Leukotriene synthases and the receptors induced by peripheral nerve injury in the spinal cord contribute to the generation of neuropathic pain. Glia. 2009, 58 (5): 599-610.
Rashid MH, Inoue M, Matsumoto M, Ueda H: Switching of bradykinin-mediated nociception following partial sciatic nerve injury in mice. J Pharmacol Exp Ther. 2004, 308: 1158-1164. 10.1124/jpet.103.060335.
Petcu M, Dias JP, Ongali B, Thibault G, Neugebauer W, Couture R: Role of kinin B1 and B2 receptors in a rat model of neuropathic pain. Int Immunopharmacol. 2008, 8: 188-196. 10.1016/j.intimp.2007.09.009.
Berrocoso E, De Benito MD, Mico JA: Role of serotonin 5-HT1A and opioid receptors in the antiallodynic effect of tramadol in the chronic constriction injury model of neuropathic pain in rats. Psychopharmacology (Berl). 2007, 193: 97-105. 10.1007/s00213-007-0761-8.
Faerber L, Drechsler S, Ladenburger S, Gschaidmeier H, Fischer W: The neuronal 5-HT3 receptor network after 20 years of research--evolving concepts in management of pain and inflammation. Eur J Pharmacol. 2007, 560: 1-8. 10.1016/j.ejphar.2007.01.028.
Rahman W, Suzuki R, Webber M, Hunt SP, Dickenson AH: Depletion of endogenous spinal 5-HT attenuates the behavioural hypersensitivity to mechanical and cooling stimuli induced by spinal nerve ligation. Pain. 2006, 123: 264-274. 10.1016/j.pain.2006.02.033.
Inoue K: P2 receptors and chronic pain. Purinergic Signal. 2007, 3: 135-144. 10.1007/s11302-006-9045-8.
Inoue K, Tsuda M, Koizumi S: ATP- and adenosine-mediated signaling in the central nervous system: chronic pain and microglia: involvement of the ATP receptor P2X4. J Pharmacol Sci. 2004, 94: 112-114. 10.1254/jphs.94.112.
Inoue K, Tsuda M, Tozaki-Saitoh H: Modification of neuropathic pain sensation through microglial ATP receptors. Purinergic Signal. 2007, 3: 311-316. 10.1007/s11302-007-9071-1.
Wilson-Gerwing TD, Stucky CL, McComb GW, Verge VM: Neurotrophin-3 significantly reduces sodium channel expression linked to neuropathic pain states. Exp Neurol. 2008, 213: 303-314. 10.1016/j.expneurol.2008.06.002.
Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y: BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005, 438: 1017-1021. 10.1038/nature04223.
Boudes M, Menigoz A: Non-neuronal BDNF, a key player in development of central sensitization and neuropathic pain. J Physiol. 2009, 587: 2111-2112. 10.1113/jphysiol.2009.172130.
Chien CC, Fu WM, Huang HI, Lai YH, Tsai YF, Guo SL, Wu TJ, Ling QD: Expression of neurotrophic factors in neonatal rats after peripheral inflammation. J Pain. 2007, 8: 161-167. 10.1016/j.jpain.2006.07.004.
Sommer C, Kress M: Recent findings on how proinflammatory cytokines cause pain: peripheral mechanisms in inflammatory and neuropathic hyperalgesia. Neurosci Lett. 2004, 361: 184-187. 10.1016/j.neulet.2003.12.007.
Okamoto K, Martin DP, Schmelzer JD, Mitsui Y, Low PA: Pro- and anti-inflammatory cytokine gene expression in rat sciatic nerve chronic constriction injury model of neuropathic pain. Exp Neurol. 2001, 169: 386-391. 10.1006/exnr.2001.7677.
Arruda JL, Colburn RW, Rickman AJ, Rutkowski MD, DeLeo JA: Increase of interleukin-6 mRNA in the spinal cord following peripheral nerve injury in the rat: potential role of IL-6 in neuropathic pain. Brain Res Mol Brain Res. 1998, 62: 228-235. 10.1016/S0169-328X(98)00257-5.
Sommer C, Schmidt C, George A: Hyperalgesia in experimental neuropathy is dependent on the TNF receptor 1. Exp Neurol. 1998, 151: 138-142. 10.1006/exnr.1998.6797.
Watkins LR, Maier SF, Goehler LE: Immune activation: the role of pro-inflammatory cytokines in inflammation, illness responses and pathological pain states. Pain. 1995, 63: 289-302. 10.1016/0304-3959(95)00186-7.
Latremoliere A, Mauborgne A, Masson J, Bourgoin S, Kayser V, Hamon M, Pohl M: Differential implication of proinflammatory cytokine interleukin-6 in the development of cephalic versus extracephalic neuropathic pain in rats. J Neurosci. 2008, 28: 8489-8501. 10.1523/JNEUROSCI.2552-08.2008.
White FA, Jung H, Miller RJ: Chemokines and the pathophysiology of neuropathic pain. Proc Natl Acad Sci USA. 2007, 104: 20151-20158. 10.1073/pnas.0709250104.
Ma W, Quirion R: Inflammatory mediators modulating the transient receptor potential vanilloid 1 receptor: therapeutic targets to treat inflammatory and neuropathic pain. Expert Opin Ther Targets. 2007, 11: 307-320. 10.1517/14728222.11.3.307.
Gao X, Kim HK, Chung JM, Chung K: Reactive oxygen species (ROS) are involved in enhancement of NMDA-receptor phosphorylation in animal models of pain. Pain. 2007, 131: 262-271. 10.1016/j.pain.2007.01.011.
Siniscalco D, Fuccio C, Giordano C, Ferraraccio F, Palazzo E, Luongo L, Rossi F, Roth KA, Maione S, de Novellis V: Role of reactive oxygen species and spinal cord apoptotic genes in the development of neuropathic pain. Pharmacol Res. 2007, 55: 158-166. 10.1016/j.phrs.2006.11.009.
Bergsteinsdottir K, Kingston A, Jessen KR: Rat Schwann cells can be induced to express major histocompatibility complex class II molecules in vivo. J Neurocytol. 1992, 21: 382-390. 10.1007/BF01191706.
Constable AL, Armati PJ, Toyka KV, Hartung HP: Production of prostanoids by Lewis rat Schwann cells in vitro. Brain Res. 1994, 635: 75-80. 10.1016/0006-8993(94)91425-7.
Bolin LM, Verity AN, Silver JE, Shooter EM, Abrams JS: Interleukin-6 production by Schwann cells and induction in sciatic nerve injury. J Neurochem. 1995, 64: 850-858.
Adler JE, Nico L, VandeVord P, Skoff AM: Modulation of neuropathic pain by a glial-derived factor. Pain Med. 2009, 10: 1229-1236. 10.1111/j.1526-4637.2009.00708.x.
Hansson E: Could chronic pain and spread of pain sensation be induced and maintained by glial activation?. Acta Physiol (Oxf). 2006, 187: 321-327. 10.1111/j.1748-1716.2006.01568.x.
Moss A, Beggs S, Vega-Avelaira D, Costigan M, Hathway GJ, Salter MW, Fitzgerald M: Spinal microglia and neuropathic pain in young rats. Pain. 2007, 128: 215-224. 10.1016/j.pain.2006.09.018.
Thacker MA, Clark AK, Bishop T, Grist J, Yip PK, Moon LD, Thompson SW, Marchand F, McMahon SB: CCL2 is a key mediator of microglia activation in neuropathic pain states. Eur J Pain. 2009, 13: 263-272. 10.1016/j.ejpain.2008.04.017.
Sauder DN: The role of epidermal cytokines in inflammatory skin diseases. J Invest Dermatol. 1990, 95: 27S-28S. 10.1111/1523-1747.ep12505705.
Compston A, Zajicek J, Sussman J, Webb A, Hall G, Muir D, Shaw C, Wood A, Scolding N: Glial lineages and myelination in the central nervous system. J Anat. 1997, 190 (Pt 2): 161-200. 10.1046/j.1469-7580.1997.19020161.x.
Lisak RP, Skundric D, Bealmear B, Ragheb S: The role of cytokines in Schwann cell damage, protection, and repair. J Infect Dis. 1997, 176 (Suppl 2): S173-179. 10.1086/513788.
Beeson PB: Temperature-elevating effect of a substance obtained from polymorphonuclear leucocytes. J Clin Invest. 1948, 27: 524.
Uceyler N, Sommer C: Cytokine regulation in animal models of neuropathic pain and in human diseases. Neurosci Lett. 2008, 437: 194-198. 10.1016/j.neulet.2008.03.050.
Wagner R, Myers RR: Endoneurial injection of TNF-alpha produces neuropathic pain behaviors. Neuroreport. 1996, 7: 2897-2901. 10.1097/00001756-199611250-00018.
Wagner R, Myers RR: Schwann cells produce tumor necrosis factor alpha: expression in injured and non-injured nerves. Neuroscience. 1996, 73: 625-629. 10.1016/0306-4522(96)00127-3.
Zelenka M, Schafers M, Sommer C: Intraneural injection of interleukin-1beta and tumor necrosis factor-alpha into rat sciatic nerve at physiological doses induces signs of neuropathic pain. Pain. 2005, 116: 257-263. 10.1016/j.pain.2005.04.018.
Sweitzer SM, Colburn RW, Rutkowski M, DeLeo JA: Acute peripheral inflammation induces moderate glial activation and spinal IL-1beta expression that correlates with pain behavior in the rat. Brain Res. 1999, 829: 209-221. 10.1016/S0006-8993(99)01326-8.
DeLeo JA, Colburn RW, Nichols M, Malhotra A: Interleukin-6-mediated hyperalgesia/allodynia and increased spinal IL-6 expression in a rat mononeuropathy model. J Interferon Cytokine Res. 1996, 16: 695-700. 10.1089/jir.1996.16.695.
Wells MR, Racis SP, Vaidya U: Changes in plasma cytokines associated with peripheral nerve injury. J Neuroimmunol. 1992, 39: 261-268. 10.1016/0165-5728(92)90260-R.
Bickels J, Kollender Y, Merinsky O, Meller I: Coley's toxin: historical perspective. Isr Med Assoc J. 2002, 4: 471-472.
Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B: An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci USA. 1975, 72: 3666-3670. 10.1073/pnas.72.9.3666.
Bodmer JL, Schneider P, Tschopp J: The molecular architecture of the TNF superfamily. Trends Biochem Sci. 2002, 27: 19-26. 10.1016/S0968-0004(01)01995-8.
Locksley RM, Killeen N, Lenardo MJ: The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001, 104: 487-501. 10.1016/S0092-8674(01)00237-9.
George A, Schmidt C, Weishaupt A, Toyka KV, Sommer C: Serial determination of tumor necrosis factor-alpha content in rat sciatic nerve after chronic constriction injury. Exp Neurol. 1999, 160: 124-132. 10.1006/exnr.1999.7193.
George A, Buehl A, Sommer C: Wallerian degeneration after crush injury of rat sciatic nerve increases endo- and epineurial tumor necrosis factor-alpha protein. Neurosci Lett. 2004, 372: 215-219. 10.1016/j.neulet.2004.09.075.
Shubayev VI, Myers RR: Upregulation and interaction of TNFalpha and gelatinases A and B in painful peripheral nerve injury. Brain Res. 2000, 855: 83-89. 10.1016/S0006-8993(99)02321-5.
Sommer C, Schafers M: Painful mononeuropathy in C57BL/Wld mice with delayed wallerian degeneration: differential effects of cytokine production and nerve regeneration on thermal and mechanical hypersensitivity. Brain Res. 1998, 784: 154-162. 10.1016/S0006-8993(97)01327-9.
Shamash S, Reichert F, Rotshenker S: The cytokine network of Wallerian degeneration: tumor necrosis factor-alpha, interleukin-1alpha, and interleukin-1beta. J Neurosci. 2002, 22: 3052-3060.
George A, Buehl A, Sommer C: Tumor necrosis factor receptor 1 and 2 proteins are differentially regulated during Wallerian degeneration of mouse sciatic nerve. Exp Neurol. 2005, 192: 163-166. 10.1016/j.expneurol.2004.11.002.
Empl M, Renaud S, Erne B, Fuhr P, Straube A, Schaeren-Wiemers N, Steck AJ: TNF-alpha expression in painful and nonpainful neuropathies. Neurology. 2001, 56: 1371-1377.
Sorkin LS, Doom CM: Epineurial application of TNF elicits an acute mechanical hyperalgesia in the awake rat. J Peripher Nerv Syst. 2000, 5: 96-100. 10.1046/j.1529-8027.2000.00012.x.
Jin X, Gereau RWt: Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J Neurosci. 2006, 26: 246-255. 10.1523/JNEUROSCI.3858-05.2006.
Yang L, Lindholm K, Konishi Y, Li R, Shen Y: Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival through different signal transduction pathways. J Neurosci. 2002, 22: 3025-3032.
Constantin CE, Mair N, Sailer CA, Andratsch M, Xu ZZ, Blumer MJ, Scherbakov N, Davis JB, Bluethmann H, Ji RR, Kress M: Endogenous tumor necrosis factor alpha (TNFalpha) requires TNF receptor type 2 to generate heat hyperalgesia in a mouse cancer model. J Neurosci. 2008, 28: 5072-5081. 10.1523/JNEUROSCI.4476-07.2008.
Schafers M, Sommer C, Geis C, Hagenacker T, Vandenabeele P, Sorkin LS: Selective stimulation of either tumor necrosis factor receptor differentially induces pain behavior in vivo and ectopic activity in sensory neurons in vitro. Neuroscience. 2008, 157: 414-423. 10.1016/j.neuroscience.2008.08.067.
Ohishi S: [Evaluation of time course and inter-relationship of inflammatory mediators in experimental inflammatory reaction]. Yakugaku Zasshi. 2000, 120: 455-462.
Tonini G, Santini D, Vincenzi B, Borzomati D, Dicuonzo G, La Cesa A, Onori N, Coppola R: Oxaliplatin may induce cytokine-release syndrome in colorectal cancer patients. J Biol Regul Homeost Agents. 2002, 16: 105-109.
Drory VE, Lev D, Groozman GB, Gutmann M, Klausner JM: Neurotoxicity of isolated limb perfusion with tumor necrosis factor. J Neurol Sci. 1998, 158: 1-4. 10.1016/S0022-510X(98)00098-7.
Schafers M, Geis C, Svensson CI, Luo ZD, Sommer C: Selective increase of tumour necrosis factor-alpha in injured and spared myelinated primary afferents after chronic constrictive injury of rat sciatic nerve. Eur J Neurosci. 2003, 17: 791-804. 10.1046/j.1460-9568.2003.02504.x.
Jancalek R, Dubovy P, Svizenska I, Klusakova I: Bilateral changes of TNF-alpha and IL-10 protein in the lumbar and cervical dorsal root ganglia following a unilateral chronic constriction injury of the sciatic nerve. J Neuroinflammation. 2010, 7: 11-10.1186/1742-2094-7-11.
Shubayev VI, Myers RR: Axonal transport of TNF-alpha in painful neuropathy: distribution of ligand tracer and TNF receptors. J Neuroimmunol. 2001, 114: 48-56. 10.1016/S0165-5728(00)00453-7.
Schafers M, Sorkin LS, Geis C, Shubayev VI: Spinal nerve ligation induces transient upregulation of tumor necrosis factor receptors 1 and 2 in injured and adjacent uninjured dorsal root ganglia in the rat. Neurosci Lett. 2003, 347: 179-182. 10.1016/S0304-3940(03)00695-5.
Sacerdote P, Franchi S, Trovato AE, Valsecchi AE, Panerai AE, Colleoni M: Transient early expression of TNF-alpha in sciatic nerve and dorsal root ganglia in a mouse model of painful peripheral neuropathy. Neurosci Lett. 2008, 436: 210-213. 10.1016/j.neulet.2008.03.023.
Cuellar JM, Montesano PX, Carstens E: Role of TNF-alpha in sensitization of nociceptive dorsal horn neurons induced by application of nucleus pulposus to L5 dorsal root ganglion in rats. Pain. 2004, 110: 578-587. 10.1016/j.pain.2004.03.029.
Shubayev VI, Myers RR: Anterograde TNF alpha transport from rat dorsal root ganglion to spinal cord and injured sciatic nerve. Neurosci Lett. 2002, 320: 99-101. 10.1016/S0304-3940(02)00010-1.
Schafers M, Lee DH, Brors D, Yaksh TL, Sorkin LS: Increased sensitivity of injured and adjacent uninjured rat primary sensory neurons to exogenous tumor necrosis factor-alpha after spinal nerve ligation. J Neurosci. 2003, 23: 3028-3038.
Thorburn A: Death receptor-induced cell killing. Cell Signal. 2004, 16: 139-144. 10.1016/j.cellsig.2003.08.007.
Micheau O, Tschopp J: Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003, 114: 181-190. 10.1016/S0092-8674(03)00521-X.
Joseph EK, Levine JD: Caspase signalling in neuropathic and inflammatory pain in the rat. Eur J Neurosci. 2004, 20: 2896-2902. 10.1111/j.1460-9568.2004.03750.x.
Sekiguchi M, Sekiguchi Y, Konno S, Kobayashi H, Homma Y, Kikuchi S: Comparison of neuropathic pain and neuronal apoptosis following nerve root or spinal nerve compression. Eur Spine J. 2009, 18: 1978-1985. 10.1007/s00586-009-1064-z.
Czeschik JC, Hagenacker T, Schafers M, Busselberg D: TNF-alpha differentially modulates ion channels of nociceptive neurons. Neurosci Lett. 2008, 434: 293-298. 10.1016/j.neulet.2008.01.070.
Watkins LR, Maier SF: The pain of being sick: implications of immune-to-brain communication for understanding pain. Annu Rev Psychol. 2000, 51: 29-57. 10.1146/annurev.psych.51.1.29.
Ignatowski TA, Covey WC, Knight PR, Severin CM, Nickola TJ, Spengler RN: Brain-derived TNFalpha mediates neuropathic pain. Brain Res. 1999, 841: 70-77. 10.1016/S0006-8993(99)01782-5.
Covey WC, Ignatowski TA, Knight PR, Spengler RN: Brain-derived TNFalpha: involvement in neuroplastic changes implicated in the conscious perception of persistent pain. Brain Res. 2000, 859: 113-122. 10.1016/S0006-8993(00)01965-X.
Covey WC, Ignatowski TA, Renauld AE, Knight PR, Nader ND, Spengler RN: Expression of neuron-associated tumor necrosis factor alpha in the brain is increased during persistent pain. Reg Anesth Pain Med. 2002, 27: 357-366.
Li X, Wang J, Wang Z, Dong C, Dong X, Jing Y, Yuan Y, Fan G: Tumor necrosis factor-alpha of Red nucleus involved in the development of neuropathic allodynia. Brain Res Bull. 2008.
Sanchez I, Hassinger L, Paskevich PA, Shine HD, Nixon RA: Oligodendroglia regulate the regional expansion of axon caliber and local accumulation of neurofilaments during development independently of myelin formation. J Neurosci. 1996, 16: 5095-5105.
Nadkarni S, Jung P: Modeling synaptic transmission of the tripartite synapse. Phys Biol. 2007, 4: 1-9. 10.1088/1478-3975/4/1/001.
Stevens B: Neuron-astrocyte signaling in the development and plasticity of neural circuits. Neurosignals. 2008, 16: 278-288. 10.1159/000123038.
Haydon PG, Carmignoto G: Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev. 2006, 86: 1009-1031. 10.1152/physrev.00049.2005.
Ji RR, Kawasaki Y, Zhuang ZY, Wen YR, Decosterd I: Possible role of spinal astrocytes in maintaining chronic pain sensitization: review of current evidence with focus on bFGF/JNK pathway. Neuron Glia Biol. 2006, 2: 259-269. 10.1017/S1740925X07000403.
Ji RR, Suter MR: p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain. 2007, 3: 33-10.1186/1744-8069-3-33.
De Leo JA, Tawfik VL, LaCroix-Fralish ML: The tetrapartite synapse: path to CNS sensitization and chronic pain. Pain. 2006, 122: 17-21. 10.1016/j.pain.2006.02.034.
Seth P, Koul N: Astrocyte, the star avatar: redefined. J Biosci. 2008, 33: 405-421. 10.1007/s12038-008-0060-5.
Vilhardt F: Microglia: phagocyte and glia cell. Int J Biochem Cell Biol. 2005, 37: 17-21. 10.1016/j.biocel.2004.06.010.
Watkins LR, Maier SF: Glia: a novel drug discovery target for clinical pain. Nat Rev Drug Discov. 2003, 2: 973-985. 10.1038/nrd1251.
Ren K, Dubner R: Neuron-glia crosstalk gets serious: role in pain hypersensitivity. Curr Opin Anaesthesiol. 2008, 21: 570-579. 10.1097/ACO.0b013e32830edbdf.
Abbadie C, Bhangoo S, De Koninck Y, Malcangio M, Melik-Parsadaniantz S, White FA: Chemokines and pain mechanisms. Brain Res Rev. 2008, 60 (1): 125-34. 10.1016/j.brainresrev.2008.12.002.
Inoue K, Tsuda M, Koizumi S: Chronic pain and microglia: the role of ATP. Novartis Found Symp. 2004, 261: 55-64. full_text. discussion 64-57, 149-154
Tsuda M, Inoue K: [Neuropathic pain and ATP receptors in spinal microglia]. Brain Nerve. 2007, 59: 953-959.
Garrison CJ, Dougherty PM, Kajander KC, Carlton SM: Staining of glial fibrillary acidic protein (GFAP) in lumbar spinal cord increases following a sciatic nerve constriction injury. Brain Res. 1991, 565: 1-7. 10.1016/0006-8993(91)91729-K.
Watkins LR, Milligan ED, Maier SF: Glial activation: a driving force for pathological pain. Trends Neurosci. 2001, 24: 450-455. 10.1016/S0166-2236(00)01854-3.
Watkins LR, Hutchinson MR, Milligan ED, Maier SF: "Listening" and "talking" to neurons: implications of immune activation for pain control and increasing the efficacy of opioids. Brain Res Rev. 2007, 56: 148-169. 10.1016/j.brainresrev.2007.06.006.
Wei F, Guo W, Zou S, Ren K, Dubner R: Supraspinal glial-neuronal interactions contribute to descending pain facilitation. J Neurosci. 2008, 28: 10482-10495. 10.1523/JNEUROSCI.3593-08.2008.
Bliss TV, Collingridge GL: A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993, 361: 31-39. 10.1038/361031a0.
Rygh LJ, Svendsen F, Fiska A, Haugan F, Hole K, Tjolsen A: Long-term potentiation in spinal nociceptive systems--how acute pain may become chronic. Psychoneuroendocrinology. 2005, 30: 959-964. 10.1016/j.psyneuen.2005.04.007.
Ikeda H, Kiritoshi T, Murase K: Synaptic plasticity in the spinal dorsal horn. Neurosci Res. 2009, 64: 133-136. 10.1016/j.neures.2009.03.004.
Liu XG, Sandkuhler J: Long-term potentiation of C-fiber-evoked potentials in the rat spinal dorsal horn is prevented by spinal N-methyl-D-aspartic acid receptor blockage. Neurosci Lett. 1995, 191: 43-46. 10.1016/0304-3940(95)11553-0.
Liu X, Sandkuhler J: Characterization of long-term potentiation of C-fiber-evoked potentials in spinal dorsal horn of adult rat: essential role of NK1 and NK2 receptors. J Neurophysiol. 1997, 78: 1973-1982.
Zhang HM, Zhou LJ, Hu XD, Hu NW, Zhang T, Liu XG: Acute nerve injury induces long-term potentiation of C-fiber evoked field potentials in spinal dorsal horn of intact rat. Sheng Li Xue Bao. 2004, 56: 591-596.
Klein T, Magerl W, Hopf HC, Sandkuhler J, Treede RD: Perceptual correlates of nociceptive long-term potentiation and long-term depression in humans. J Neurosci. 2004, 24: 964-971. 10.1523/JNEUROSCI.1222-03.2004.
Klein T, Magerl W, Treede RD: Perceptual correlate of nociceptive long-term potentiation (LTP) in humans shares the time course of early-LTP. J Neurophysiol. 2006, 96: 3551-3555. 10.1152/jn.00755.2006.
Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC: Control of synaptic strength by glial TNFalpha. Science. 2002, 295: 2282-2285. 10.1126/science.1067859.
Stellwagen D, Malenka RC: Synaptic scaling mediated by glial TNF-alpha. Nature. 2006, 440: 1054-1059. 10.1038/nature04671.
Pickering M, Cumiskey D, O'Connor JJ: Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp Physiol. 2005, 90: 663-670. 10.1113/expphysiol.2005.030734.
Tancredi V, D'Arcangelo G, Grassi F, Tarroni P, Palmieri G, Santoni A, Eusebi F: Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neurosci Lett. 1992, 146: 176-178. 10.1016/0304-3940(92)90071-E.
Liu YL, Zhou LJ, Hu NW, Xu JT, Wu CY, Zhang T, Li YY, Liu XG: Tumor necrosis factor-alpha induces long-term potentiation of C-fiber evoked field potentials in spinal dorsal horn in rats with nerve injury: the role of NF-kappa B, JNK and p38 MAPK. Neuropharmacology. 2007, 52: 708-715. 10.1016/j.neuropharm.2006.09.011.
Holton FA, Holton P: The capillary dilator substances in dry powders of spinal roots; a possible role of adenosine triphosphate in chemical transmission from nerve endings. J Physiol. 1954, 126: 124-140.
Holton P: The liberation of adenosine triphosphate on antidromic stimulation of sensory nerves. J Physiol. 1959, 145: 494-504.
Bleehen T, Keele CA: Observations on the algogenic actions of adenosine compounds on the human blister base preparation. Pain. 1977, 3: 367-377. 10.1016/0304-3959(77)90066-5.
Bleehen T: The effects of adenine nucleotides on cutaneous afferent nerve activity. Br J Pharmacol. 1978, 62: 573-577.
Jahr CE, Jessell TM: ATP excites a subpopulation of rat dorsal horn neurones. Nature. 1983, 304: 730-733. 10.1038/304730a0.
Burnstock G: Purinergic nerves and receptors. Prog Biochem Pharmacol. 1980, 16: 141-154.
Burnstock G, Kennedy C: Is there a basis for distinguishing two types of P2-purinoceptor?. Gen Pharmacol. 1985, 16: 433-440.
Burnstock G: Purinergic signalling and disorders of the central nervous system. Nat Rev Drug Discov. 2008, 7: 575-590. 10.1038/nrd2605.
Burnstock G: Physiology and pathophysiology of purinergic neurotransmission. Physiol Rev. 2007, 87: 659-797. 10.1152/physrev.00043.2006.
Amaya F, Wang H, Costigan M, Allchorne AJ, Hatcher JP, Egerton J, Stean T, Morisset V, Grose D, Gunthorpe MJ, et al: The voltage-gated sodium channel Na(v)1.9 is an effector of peripheral inflammatory pain hypersensitivity. J Neurosci. 2006, 26: 12852-12860. 10.1523/JNEUROSCI.4015-06.2006.
Novakovic SD, Kassotakis LC, Oglesby IB, Smith JA, Eglen RM, Ford AP, Hunter JC: Immunocytochemical localization of P2X3 purinoceptors in sensory neurons in naive rats and following neuropathic injury. Pain. 1999, 80: 273-282. 10.1016/S0304-3959(98)00225-5.
Bradbury EJ, Burnstock G, McMahon SB: The expression of P2X3 purinoreceptors in sensory neurons: effects of axotomy and glial-derived neurotrophic factor. Mol Cell Neurosci. 1998, 12: 256-268. 10.1006/mcne.1998.0719.
Barclay J, Patel S, Dorn G, Wotherspoon G, Moffatt S, Eunson L, Abdel'al S, Natt F, Hall J, Winter J, et al: Functional downregulation of P2X3 receptor subunit in rat sensory neurons reveals a significant role in chronic neuropathic and inflammatory pain. J Neurosci. 2002, 22: 8139-8147.
Dorn G, Patel S, Wotherspoon G, Hemmings-Mieszczak M, Barclay J, Natt FJ, Martin P, Bevan S, Fox A, Ganju P, et al: siRNA relieves chronic neuropathic pain. Nucleic Acids Res. 2004, 32: e49-10.1093/nar/gnh044.
Honore P, Kage K, Mikusa J, Watt AT, Johnston JF, Wyatt JR, Faltynek CR, Jarvis MF, Lynch K: Analgesic profile of intrathecal P2X(3) antisense oligonucleotide treatment in chronic inflammatory and neuropathic pain states in rats. Pain. 2002, 99: 11-19. 10.1016/S0304-3959(02)00032-5.
Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K: P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003, 424: 778-783. 10.1038/nature01786.
Ulmann L, Hatcher JP, Hughes JP, Chaumont S, Green PJ, Conquet F, Buell GN, Reeve AJ, Chessell IP, Rassendren F: Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J Neurosci. 2008, 28: 11263-11268. 10.1523/JNEUROSCI.2308-08.2008.
Trang T, Beggs S, Salter MW: Purinoceptors in microglia and neuropathic pain. Pflugers Arch. 2006, 452: 645-652. 10.1007/s00424-006-0074-5.
Gong QJ, Li YY, Xin WJ, Zang Y, Ren WJ, Wei XH, Zhang T, Liu XG: ATP induces long-term potentiation of C-fiber-evoked field potentials in spinal dorsal horn: the roles of P2X4 receptors and p38 MAPK in microglia. Glia. 2009, 57: 583-591. 10.1002/glia.20786.
Suzuki T, Hide I, Ido K, Kohsaka S, Inoue K, Nakata Y: Production and release of neuroprotective tumor necrosis factor by P2X7 receptor-activated microglia. J Neurosci. 2004, 24: 1-7. 10.1523/JNEUROSCI.3792-03.2004.
Lister MF, Sharkey J, Sawatzky DA, Hodgkiss JP, Davidson DJ, Rossi AG, Finlayson K: The role of the purinergic P2X7 receptor in inflammation. J Inflamm (Lond). 2007, 4: 5-10.1186/1476-9255-4-5.
Terayama R, Omura S, Fujisawa N, Yamaai T, Ichikawa H, Sugimoto T: Activation of microglia and p38 mitogen-activated protein kinase in the dorsal column nucleus contributes to tactile allodynia following peripheral nerve injury. Neuroscience. 2008, 153: 1245-1255. 10.1016/j.neuroscience.2008.03.041.
Wen YR, Suter MR, Ji RR, Yeh GC, Wu YS, Wang KC, Kohno T, Sun WZ, Wang CC: Activation of p38 mitogen-activated protein kinase in spinal microglia contributes to incision-induced mechanical allodynia. Anesthesiology. 2009, 110: 155-165. 10.1097/ALN.0b013e318190bc16.
Crown ED, Ye Z, Johnson KM, Xu GY, McAdoo DJ, Hulsebosch CE: Increases in the activated forms of ERK 1/2, p38 MAPK, and CREB are correlated with the expression of at-level mechanical allodynia following spinal cord injury. Exp Neurol. 2006, 199: 397-407. 10.1016/j.expneurol.2006.01.003.
Crown ED, Gwak YS, Ye Z, Johnson KM, Hulsebosch CE: Activation of p38 MAP kinase is involved in central neuropathic pain following spinal cord injury. Exp Neurol. 2008, 213: 257-267. 10.1016/j.expneurol.2008.05.025.
Schafers M, Svensson CI, Sommer C, Sorkin LS: Tumor necrosis factor-alpha induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci. 2003, 23: 2517-2521.
Sun M, Fink PJ: A new class of reverse signaling costimulators belongs to the TNF family. J Immunol. 2007, 179: 4307-4312.
Hao S, Mata M, Glorioso JC, Fink DJ: Gene transfer to interfere with TNFalpha signaling in neuropathic pain. Gene Ther. 2007, 14: 1010-1016. 10.1038/sj.gt.3302950.
Mata M, Hao S, Fink DJ: Gene therapy directed at the neuroimmune component of chronic pain with particular attention to the role of TNF alpha. Neurosci Lett. 2008, 437: 209-213. 10.1016/j.neulet.2008.03.049.
Shamji MF, Chen J, Friedman AH, Richardson WJ, Chilkoti A, Setton LA: Synthesis and characterization of a thermally-responsive tumor necrosis factor antagonist. J Control Release. 2008, 129: 179-186. 10.1016/j.jconrel.2008.04.021.
Shamji MF, Jing L, Chen J, Hwang P, Ghodsizadeh O, Friedman AH, Richardson WJ, Setton LA: Treatment of neuroinflammation by soluble tumor necrosis factor receptor Type II fused to a thermally responsive carrier. J Neurosurg Spine. 2008, 9: 221-228. 10.3171/SPI/2008/9/7/010.
Sommer C, Lindenlaub T, Teuteberg P, Schafers M, Hartung T, Toyka KV: Anti-TNF-neutralizing antibodies reduce pain-related behavior in two different mouse models of painful mononeuropathy. Brain Res. 2001, 913: 86-89. 10.1016/S0006-8993(01)02743-3.
Lindenlaub T, Teuteberg P, Hartung T, Sommer C: Effects of neutralizing antibodies to TNF-alpha on pain-related behavior and nerve regeneration in mice with chronic constriction injury. Brain Res. 2000, 866: 15-22. 10.1016/S0006-8993(00)02190-9.
Sommer C, Schafers M, Marziniak M, Toyka KV: Etanercept reduces hyperalgesia in experimental painful neuropathy. J Peripher Nerv Syst. 2001, 6: 67-72. 10.1046/j.1529-8027.2001.01010.x.
Svensson CI, Schafers M, Jones TL, Powell H, Sorkin LS: Spinal blockade of TNF blocks spinal nerve ligation-induced increases in spinal P-p38. Neurosci Lett. 2005, 379: 209-213. 10.1016/j.neulet.2004.12.064.
Marchand F, Tsantoulas C, Singh D, Grist J, Clark AK, Bradbury EJ, McMahon SB: Effects of Etanercept and Minocycline in a rat model of spinal cord injury. Eur J Pain. 2009, 13: 673-681. 10.1016/j.ejpain.2008.08.001.
Si Q, Nakamura Y, Ogata T, Kataoka K, Schubert P: Differential regulation of microglial activation by propentofylline via cAMP signaling. Brain Res. 1998, 812: 97-104. 10.1016/S0006-8993(98)00954-8.
Sweitzer SM, Schubert P, DeLeo JA: Propentofylline, a glial modulating agent, exhibits antiallodynic properties in a rat model of neuropathic pain. J Pharmacol Exp Ther. 2001, 297: 1210-1217.
Tawfik VL, Nutile-McMenemy N, Lacroix-Fralish ML, Deleo JA: Efficacy of propentofylline, a glial modulating agent, on existing mechanical allodynia following peripheral nerve injury. Brain Behav Immun. 2007, 21: 238-246. 10.1016/j.bbi.2006.07.001.
Mielke R, Moller HJ, Erkinjuntti T, Rosenkranz B, Rother M, Kittner B: Propentofylline in the treatment of vascular dementia and Alzheimer-type dementia: overview of phase I and phase II clinical trials. Alzheimer Dis Assoc Disord. 1998, 12 (Suppl 2): S29-35.
Frampton M, Harvey RJ, Kirchner V: Propentofylline for dementia. Cochrane Database Syst Rev. 2003, CD002853.
Bauditz J, Wedel S, Lochs H: Thalidomide reduces tumour necrosis factor alpha and interleukin 12 production in patients with chronic active Crohn's disease. Gut. 2002, 50: 196-200. 10.1136/gut.50.2.196.
Moreira AL, Sampaio EP, Zmuidzinas A, Frindt P, Smith KA, Kaplan G: Thalidomide exerts its inhibitory action on tumor necrosis factor alpha by enhancing mRNA degradation. J Exp Med. 1993, 177: 1675-1680. 10.1084/jem.177.6.1675.
Sommer C, Marziniak M, Myers RR: The effect of thalidomide treatment on vascular pathology and hyperalgesia caused by chronic constriction injury of rat nerve. Pain. 1998, 74: 83-91. 10.1016/S0304-3959(97)00154-1.
George A, Marziniak M, Schafers M, Toyka KV, Sommer C: Thalidomide treatment in chronic constrictive neuropathy decreases endoneurial tumor necrosis factor-alpha, increases interleukin-10 and has long-term effects on spinal cord dorsal horn met-enkephalin. Pain. 2000, 88: 267-275. 10.1016/S0304-3959(00)00333-X.
Mackey S, Feinberg S: Pharmacologic therapies for complex regional pain syndrome. Curr Pain Headache Rep. 2007, 11: 38-43. 10.1007/s11916-007-0020-z.
Goli V: Does thalidomide have an analgesic effect? Current status and future directions. Curr Pain Headache Rep. 2007, 11: 109-114. 10.1007/s11916-007-0007-9.
Clemmensen OJ, Olsen PZ, Andersen KE: Thalidomide neurotoxicity. Arch Dermatol. 1984, 120: 338-341. 10.1001/archderm.120.3.338.
el-Badawi MG, Fatani JA, Bahakim H, Abdalla MA: Light and electron microscopic observations on the cerebellum of guinea pigs following low-dose methotrexate. Exp Mol Pathol. 1990, 53: 211-222. 10.1016/0014-4800(90)90045-F.
Montesinos MC, Takedachi M, Thompson LF, Wilder TF, Fernandez P, Cronstein BN: The antiinflammatory mechanism of methotrexate depends on extracellular conversion of adenine nucleotides to adenosine by ecto-5'-nucleotidase: findings in a study of ecto-5'-nucleotidase gene-deficient mice. Arthritis Rheum. 2007, 56: 1440-1445. 10.1002/art.22643.
Montesinos MC, Yap JS, Desai A, Posadas I, McCrary CT, Cronstein BN: Reversal of the antiinflammatory effects of methotrexate by the nonselective adenosine receptor antagonists theophylline and caffeine: evidence that the antiinflammatory effects of methotrexate are mediated via multiple adenosine receptors in rat adjuvant arthritis. Arthritis Rheum. 2000, 43: 656-663. 10.1002/1529-0131(200003)43:3<656::AID-ANR23>3.0.CO;2-H.
Segal R, Mozes E, Yaron M, Tartakovsky B: The effects of methotrexate on the production and activity of interleukin-1. Arthritis Rheum. 1989, 32: 370-377. 10.1002/anr.1780320403.
Hashizume H, Rutkowski MD, Weinstein JN, DeLeo JA: Central administration of methotrexate reduces mechanical allodynia in an animal model of radiculopathy/sciatica. Pain. 2000, 87: 159-169. 10.1016/S0304-3959(00)00281-5.
Bruce-Gregorios JH, Soucy D, Chen MG, Benson N: Effect of methotrexate on cell cycle and DNA synthesis of astrocytes in primary culture: flow cytometric studies. J Neuropathol Exp Neurol. 1991, 50: 63-72. 10.1097/00005072-199101000-00005.
Bruce-Gregorios JH, Soucy DM, Chen MG, Norenberg MD: Effect of methotrexate on glial fibrillary acidic protein content of astrocytes in primary culture. J Neuropathol Exp Neurol. 1991, 50: 118-125. 10.1097/00005072-199103000-00003.
Gregorios JB, Gregorios AB, Mora J, Marcillo A, Fojaco RM, Green B: Morphologic alterations in rat brain following systemic and intraventricular methotrexate injection: light and electron microscopic studies. J Neuropathol Exp Neurol. 1989, 48: 33-47. 10.1097/00005072-198901000-00004.
Genevay S, Finckh A, Payer M, Mezin F, Tessitore E, Gabay C, Guerne PA: Elevated levels of tumor necrosis factor-alpha in periradicular fat tissue in patients with radiculopathy from herniated disc. Spine. 2008, 33: 2041-2046. 10.1097/BRS.0b013e318183bb86.
Genevay S, Stingelin S, Gabay C: Efficacy of etanercept in the treatment of acute, severe sciatica: a pilot study. Ann Rheum Dis. 2004, 63: 1120-1123. 10.1136/ard.2003.016451.
Karppinen J, Korhonen T, Malmivaara A, Paimela L, Kyllonen E, Lindgren KA, Rantanen P, Tervonen O, Niinimaki J, Seitsalo S, Hurri H: Tumor necrosis factor-alpha monoclonal antibody, infliximab, used to manage severe sciatica. Spine. 2003, 28: 750-753. 10.1097/00007632-200304150-00004. discussion 753-754
Korhonen T, Karppinen J, Malmivaara A, Autio R, Niinimaki J, Paimela L, Kyllonen E, Lindgren KA, Tervonen O, Seitsalo S, Hurri H: Efficacy of infliximab for disc herniation-induced sciatica: one-year follow-up. Spine. 2004, 29: 2115-2119. 10.1097/01.brs.0000141179.58778.6c.
Korhonen T, Karppinen J, Paimela L, Malmivaara A, Lindgren KA, Jarvinen S, Niinimaki J, Veeger N, Seitsalo S, Hurri H: The treatment of disc herniation-induced sciatica with infliximab: results of a randomized, controlled, 3-month follow-up study. Spine. 2005, 30: 2724-2728. 10.1097/01.brs.0000190815.13764.64.
Korhonen T, Karppinen J, Paimela L, Malmivaara A, Lindgren KA, Bowman C, Hammond A, Kirkham B, Jarvinen S, Niinimaki J, et al: The treatment of disc-herniation-induced sciatica with infliximab: one-year follow-up results of FIRST II, a randomized controlled trial. Spine. 2006, 31: 2759-2766. 10.1097/01.brs.0000245873.23876.1e.
Cohen SP, Wenzell D, Hurley RW, Kurihara C, Buckenmaier CC, Griffith S, Larkin TM, Dahl E, Morlando BJ: A double-blind, placebo-controlled, dose-response pilot study evaluating intradiscal etanercept in patients with chronic discogenic low back pain or lumbosacral radiculopathy. Anesthesiology. 2007, 107: 99-105. 10.1097/01.anes.0000267518.20363.0d.
Cohen SP, Bogduk N, Dragovich A, Buckenmaier CC, Griffith S, Kurihara C, Raymond J, Richter PJ, Williams N, Yaksh TL: Randomized, double-blind, placebo-controlled, dose-response, and preclinical safety study of transforaminal epidural etanercept for the treatment of sciatica. Anesthesiology. 2009, 110: 1116-1126. 10.1097/ALN.0b013e3181a05aa0.
Rolan P, Gibbons JA, He L, Chang E, Jones D, Gross MI, Davidson JB, Sanftner LM, Johnson KW: Ibudilast in healthy volunteers: safety, tolerability and pharmacokinetics with single and multiple doses. Br J Clin Pharmacol. 2008, 66: 792-801. 10.1111/j.1365-2125.2008.03270.x.
Ledeboer A, Liu T, Shumilla JA, Mahoney JH, Vijay S, Gross MI, Vargas JA, Sultzbaugh L, Claypool MD, Sanftner LM, et al: The glial modulatory drug AV411 attenuates mechanical allodynia in rat models of neuropathic pain. Neuron Glia Biol. 2006, 2: 279-291. 10.1017/S1740925X0700035X.
Watkins LR, Milligan ED, Maier SF: Glial proinflammatory cytokines mediate exaggerated pain states: implications for clinical pain. Adv Exp Med Biol. 2003, 521: 1-21.
Wieseler-Frank J, Maier SF, Watkins LR: Glial activation and pathological pain. Neurochem Int. 2004, 45: 389-395. 10.1016/j.neuint.2003.09.009.
Milligan ED, Watkins LR: Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009, 10: 23-36. 10.1038/nrn2533.
Milligan ED, Sloane EM, Watkins LR: Glia in pathological pain: a role for fractalkine. J Neuroimmunol. 2008, 198: 113-120. 10.1016/j.jneuroim.2008.04.011.
Bettoni I, Comelli F, Rossini C, Granucci F, Giagnoni G, Peri F, Costa B: Glial TLR4 receptor as new target to treat neuropathic pain: efficacy of a new receptor antagonist in a model of peripheral nerve injury in mice. Glia. 2008, 56: 1312-1319. 10.1002/glia.20699.
Schafers M, Sommer C: Anticytokine therapy in neuropathic pain management. Expert Rev Neurother. 2007, 7: 1613-1627. 10.1586/14737175.7.11.1613.
Gao YJ, Ji RR: Chemokines, neuronal-glial interactions, and central processing of neuropathic pain. Pharmacol Ther. 2010, 126 (1): 56-68. 10.1016/j.pharmthera.2010.01.002.
Ji RR, Gereau RWt, Malcangio M, Strichartz GR: MAP kinase and pain. Brain Res Rev. 2008, 60 (1): 135-48. 10.1016/j.brainresrev.2008.12.011.
Ledeboer A, Hutchinson MR, Watkins LR, Johnson KW: Ibudilast (AV-411). A new class therapeutic candidate for neuropathic pain and opioid withdrawal syndromes. Expert Opin Investig Drugs. 2007, 16: 935-950. 10.1517/13543784.16.7.935.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
LL wrote the manuscript, CMC provided comments and proof-reading. Both authors have read and approved of the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Leung, L., Cahill, C.M. TNF-α and neuropathic pain - a review. J Neuroinflammation 7, 27 (2010). https://doi.org/10.1186/1742-2094-7-27
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-2094-7-27