
Abstract
Development of various vaccines for prostate cancer (PCa) is becoming an active research area. PCa vaccines are perceived to have less toxicity compared with the available cytotoxic agents. While various immune-based strategies can elicit anti-tumour responses, DNA vaccines present increased efficacy, inducing both humoural and cellular immunity. This immune activation has been proven effective in animal models and initial clinical trials are encouraging. However, to validate the role of DNA vaccination in currently available PCa management paradigms, strong clinical evidence is still lacking. This article provides an overview of the basic principles of DNA vaccines and aims to provide a summary of preclinical and clinical trials outlining the benefits of this immunotherapy in the management of PCa.
Similar content being viewed by others


Introduction
Prostate cancer (PCa) is a major urological problem associated with significant morbidity and mortality [1]. Although the majority of PCa cases are localised to the prostate, nearly one-third of newly diagnosed patients have advanced or metastatic disease [2]. Radical prostatectomy and radical radiotherapy are potentially curative treatment options for localised PCa. For those with locally advanced or metastatic disease, the initial systemic therapy is androgen deprivation therapy. Unfortunately, nearly all patients in this group eventually progress to castration resistant PCa (CRPC). For CRPC, docetaxel based chemotherapy is used as first-line. However, this only provides modest survival benefit (2.4 months) and is associated with significant side effects [3]. Insights into the regulation of immune responses in malignancies have facilitated the emergence of novel immune-based strategies. PCa is an attractive target for vaccination due to its slow growth which can allow sufficient time for immune activation [4]. Additionally, the identification of prostate tumour associated antigens (that are recognised by T cells) has created the opportunity to develop novel immune based therapeutic approaches. Several tumour associated antigens have been identified, including prostate specific antigen (PSA), prostatic acid phosphatase (PAP), prostate stem cell antigen (PSCA) and prostate specific membrane antigen (PSMA) [5]. These antigens are largely prostate specific and their expression is strongly upregulated in PCa, both locally and at metastatic sites [6, 7]. This makes these antigens a viable target of active immunotherapy and can be used as DNA vaccines. Furthermore, PSA-specific cellular immune responses have been detected in some PCa patients and in normal individuals, suggesting that tolerance towards this antigen may be broken [8]. These observations led to development of a variety of vaccines for PCa in pre-clinical and clinical trials. These vaccines can be broadly classified as whole cell vaccines [9], protein/peptide based vaccines [10, 11], viral vector based vaccines [12], dendritic cell vaccines [13] and naked DNA vaccines [14, 15].
Like other forms of targeted therapy, cancer vaccines hold the promise of achieving cancer control without inducing overt toxicity. The focus of extensive research for PCa vaccine has led to the approval of the first therapeutic vaccine (sipuleucel-T) by the FDA [16]. The sipuleucel-T is an autologous antigen presenting cell (APC) based and antigen-targeted immunotherapeutic innovation for men with CRPC [16]. The success story of the sipuleucel-T encouraged researchers to explore other agents/strategies to activate immune system against PCa. Gene therapy (including DNA vaccines) is a realistic prospect for the treatment of prostate and other cancers, and involves the delivery of genetic information to the patient to facilitate the production of therapeutic proteins. A DNA vaccine consists of tumour specific/associated antigen and additional immune-stimulatory factors cloned into a bacterial plasmid downstream of an appropriate eukaryotic promoter for strong and stable expression. DNA vaccination can induce effective anti-tumour responses against various malignant cells and provides an attractive strategy for the management of PCa [17–19].
Why DNA vaccines?
DNA immunisation can efficiently stimulate humoural and cellular immune responses to protein antigens. This strategy has been used successfully for infectious diseases [20] and potentially can be applied for malignant conditions. There are several advantages associated with DNA vaccines:
-
Various groups have demonstrated tumour protection using DNA immunisations in several pre-clinical cancer models [21–26].
-
Gene sequences can be manipulated easily to provide multiple potential epitopes that stimulate both humoural and cellular immunity [22, 27–29].
-
DNA vaccines generally skew the immune system toward the desired T-helper1 (Th1) response. This is most likely because plasmid DNA contains unmethylated CpG motifs, which have been shown to be a very potent immunological adjuvant [30, 31].
-
DNA immunisation is safe in humans [14, 32] and can induce antigen-specific immune responses [30, 33].
-
DNA vaccines can be produced readily at a large scale [34].
Mechanisms of action
Anti-tumour immune responses
For malignant diseases, immunological therapies fall into two general categories; active and passive immunotherapy. Active immunotherapy attempts to directly elicit tumour-specific host immune responses that control or eradicate tumours. In contrast, passive immunotherapy involves the direct administration of effector molecules, such as cytokines or monoclonal antibodies. These molecules promote the development of anti-tumour responses, directly killing tumour cells, or inhibit cell invasion and angiogenesis [35]. The nature of the host immune response that can control tumour growth has been the focus of many studies. With few exceptions, the most effective anti-tumour immune responses in animal models have depended on the efficient generation of Th1 cell immunity, characterised by strong cytotoxic T lymphocyte (CTL) responses [27, 28, 36]. This is supported by observations in humans that progressive disease is characterised by an anti-tumour T-cell response [37] as well as by clinical evidence that Th1 T cells can control and eliminate metastatic disease [28, 38]. B cell effector functions are another important component of this anti-tumour immune response (see below).
Process of DNA vaccination
A typical DNA vaccine consists of a transgene that encodes the sequence of a target protein (e.g. PSA, PSMA etc.) under the control of a eukaryotic promoter. Various modalities exist for delivering such DNA to appropriate cells (see below). After uptake of the plasmid, the target protein is produced within the cell and processed into small antigenic peptides by host proteases. The peptides then enter the lumen of the endoplasmic reticulum (E.R.) by membrane-associated transporters. In the E.R., the peptides bind to Major Histocompatability Complex I (MHC I) molecules. These peptides are presented on the cell surface in the context of MHC I. Subsequently CD8+ CTL are stimulated resulting in cell-mediated immunity. CTLs cause tumour destruction through both cytolysis of malignant cells and non-cytolysis mechanisms such as cytokines production.
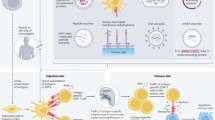
The foreign protein on the plasmid can also be presented by MHC II pathway by APC which elicit CD4+ helper T cells responses. These CD4+ T cells can recognise the peptides generated from the exogenous proteins that were endocytosed or phagocytosed by APC, then degraded to peptide fragments and loaded onto MHC II molecules. Depending on the type of CD4+ T cell that binds to the complex, B cells are activated and antibody production is stimulated. This is the same manner in which traditional vaccines work [39]. Vaccine-elicited antibodies can mediate direct effects against tumour cells by fixing complement or facilitating antibody-dependent cellular cytotoxicity. Overall, stimulation of both the T and B cell arms of the immune system mediates synergies and creates a large pool of effectors cells to control tumour growth and induce generation of memory cells (Figure 1).

Mechanism of action of DNA vaccine after intra muscular (i.m.) plasmid delivery. Transfected muscle cells produce the antigen expressed on the plasmid. This antigen is expressed to cell surface with MHC I and presented to cytotoxic CD8+ T cells (Cell mediated Immunity). Antigen is also excreted by the muscle, which is phagocytosed by the professional Antigen Presenting Cells (APC), usually Dendritic cells. A small proportion of DNA vaccine is also taken up directly by APC and the encoded antigen can then be processed and presented endogenously (Humoural Immunity).
Methods of DNA delivery
Transfection of the host cell with the plasmid is a limiting step for a successful DNA vaccination. Most commonly, the DNA vaccines are given by intramuscular (i.m.) or intradermal (i.d) injections. To facilitate the gene delivery, various methods have been reported including viral, liposomal, bacterial, ultrasound and electroporation (EP), as well as approaches involving ex vivo transfection/transduction of cells (e.g. APC) [40]. To avoid safety issues such as immune response and cytotoxicity associated with viruses and liposomal transfection, physical methods (ultrasound, EP etc.) have been widely used for either in vivo or ex vivo gene delivery. In vivo EP has emerged as a potent method for DNA vaccine delivery and significantly improves transfection efficiency of naked plasmid DNA [41]. EP driven DNA vaccination increases antigen expression by increasing transfection efficiency (Figure 2) and is accompanied by local tissue injury and inflammation [42]. Hence, the outcomes of EP mediated vaccination are dramatic enhancement of humoural and cellular immunity [43, 44].

Schematic representation of Electroporation mediated transfection. a) Intra-muscular plasmid injection. b) Electroporation. c) Transient increased permeability of cell membrane (yellow arrows) results in transfer of the plasmid into the cell. d) Cell membrane return to resting membrane (red arrow) and gene transfection results in production of mRNA and hence specific protein.
Preclinical studies
Numerous preclinical studies have been carried out to demonstrate the efficacy of DNA vaccines in animal models of PCa. The following vital issues regarding successful vaccines were addressed in these preclinical trials;
-
Induction of immune response/type of immune activation
-
Optimisation of vaccine dose/schedule
-
Mode of delivery
-
Breaking tolerance to self-antigen
-
Systemic responses /control of metastatic disease
-
Need for adjuvant to vaccines
-
Transferable immunity
Prostate-specific antigens examined
Roos et al.[45] showed for the first time that a PSA DNA vaccine can induce anti-tumour immunity in vivo. They investigated a DNA vaccination strategy to immunise mice by inducing PSA-specific cellular responses. A plasmid expressing PSA, alone or in combination with plasmids coding for granulocyte-macrophage colony-stimulating factor (GM-CSF) and/or IL-2, were used. It was found that the DNA vaccine induced PSA-specific cytotoxic T lymphocytes and when co-injected with GM-CSF and IL-2 it can protect mice against a PSA-expressing tumour challenge. This demonstrated that immunisation with a PSA DNA vaccine can evoke PSA-specific cellular immunity. The ability of the DNA vaccine to stimulate both humoural and cellular immune responses was demonstrated by Kim et al.[24]. They observed a strong and persistent antibody response against PSA for at least 180 days following immunisation. Additionally, significant T helper cell proliferation was also detected against PSA. Furthermore, immunisation with PSA plasmid induced CD8+ T cell-restricted cytotoxic T cell response against tumour cell expressing PSA.
PSMA is a 750 amino acid surface protein expressed primarily in prostate epithelium, Most reported antibodies to PSMA apparently recognise epitopes in the residue 43–570 region of the extra cellular domain, and upon binding are rapidly removed from the cell surface by internalisation. This would potentially limit their ability to mediate Fc-dependent cytotoxicity. Kuratsukuri et al.[46, 47] used a model system to target a defined region of the extra cellular domain of PSMA. Their results suggested that vaccination with plasmid expressing extra cellular domain of PSMA induced adaptive humoural activity, which was directed against the extracellular region of human PSMA and can significantly inhibit human PCa growth in athymic mice.
Viral delivery of vaccine antigens is an active research area. However, a potential difficulty with viral-based immunisations is that immune responses elicited to the viral vector might limit the possibility of multiple immunisations. Johnson et al.[48] investigated a DNA vaccine encoding PAP, to elicit antigen-specific CD8+ T cell immune responses. In their study, Lewis rats were immunised with either a plasmid DNA-based (pTVG-HP) or Vaccinia-based (VV-HP) vaccine each encoding human PAP (hPAP). They observed Th1-biased immune response (as indicated by proliferating PAP-specific CD4+/CD8+ cells and IFN- γ production) in rats immunised with a DNA vaccine encoding hPAP. Immunisation with Vaccinia virus (encoding hPAP) could not induce PAP-specific response, unless boosted with a heterologous vaccination scheme. Furthermore, they also established that multiple immunisations with a DNA vaccine encoding the rat PAP homologue (pTVG-RP) could overcome peripheral self-tolerance against rat PAP (rPAP) and generate a Th1-biased antigen-specific CD4+ and CD8+ T cell response. In separate experiments, this DNA vaccine has not shown significant toxicities in terms of animal weights, histopathology, haematological changes, or changes in serum chemistries [49]. The vaccine was found to be effective in eliciting PAP-specific CD4+ and CD8+ T cells, predominantly Th1 in type, in all immunised animals at all doses and numbers of immunisations. PAP-specific IgG were detected in a dose-dependent fashion, with titres increasing after multiple immunisations.
Optimisation of DNA vaccination
In preclinical trials different doses and vaccination schedules are investigated with variable results however, there is lack of consensus on these issues. Ahmad et al. have shown that the a DNA vaccine encoding human PSA significantly delayed the appearance of tumours and resulted in prolonged survival of the animals [50]. Additionally, a four-dose vaccination regimen provided optimal immunological effects and co-administration of synthetic CpG enhanced the tumour protective responses [50]. Furthermore, these immune responses were tumour specific and were transferable in adoptive T cell transfer experiments [50]. A DNA vaccination has the potential to break tolerance to self antigen [34, 48, 51]. A PSCA DNA vaccine when delivered by i.m. EP [51] or orally administered bacteria, [52] resulted in induction of anti-tumour immune responses against PSCA expressing subcutaneous tumours and metastatic deposits. There was activation of Th-1 type immunity against PSCA, indicating the breaking of tolerance to a self-antigen [51]. This immunity was tumour specific and was transferable by adoptive transfer of splenocytes [51].
PSMA is present in both secretary form and in prostate cells. Mincheff et al.[53] evaluated two plasmid DNA vaccines, encoding either PSMA products that are retained in the cytosol and degraded in the proteasome (tVacs; hPSMAt), or secreted proteins (sVacs; hPSMAs) for stimulation of cytotoxic cell or antibody responses. They observed that immunisation with both vectors led to generation of cell cytotoxicity, provided GM-CSF was administered with the vaccine. Spleen cells from animals immunised with hPSMAt demonstrated stronger cytotoxicity to the target cells. Interestingly, priming with a vector that encoded a xenogeneic protein (hPSMAt; ‘xenogeneic’ construct) and boosting with a vector that encoded an autologous protein (rPSMAt; ‘autologous’ construct) gave the best protection against tumour challenge. Immunisation with tVacs did not lead to formation of antibodies to the target protein, while immunisation with sVacs or with the protein did (mixed Th1-Th2 isotype). But, priming with tVacs and boosting with protein also resulted in the antibodies from the cytotoxic Th1 isotype. Hence, the best strategy to obtain a strong cellular cytotoxic response seems to be gene-based vaccinations with tVacs, priming with the ‘xenogeneic’ and boosting with the ‘autologous’ constructs.
While DNA vaccine injections, either i.m. or i.d., have been used successfully in many trials, it is still to establish which route is better. To enhance the efficacy of DNA vaccine against PCa, Roos et al.[54] have demonstrated that i.d. DNA vaccination, followed by two sets of electrical pulses of different length and voltage, can effectively induce PSA-specific T cells response. Ahmad et al. have reported successful DNA vaccination following i.m. injection coupled with EP. [50, 51] These studies indicate that EP significantly enhances transfection and hence immune activation.
Clinical trials
Preclinical studies provided encouraging evidence of enhanced immune responses and tumour protection by DNA vaccine in animal models of PCa. The success achieved in these studies resulted in exploration of application of DNA vaccine in PCa patients. To our knowledge, no naked DNA vaccine has been used in a randomised clinical trial to date [55]. However, DNA vaccines have been used in phase I/II clinical trials including patients with PCa (Table 1). Some of these clinical trials are discussed below.
In a phase I trial, Pavlenko et al.[15] investigated the feasibility, safety and immunogenicity of a DNA vaccine (pVAX/PSA) in patients with CRPC. Cytokines, GM-CSF and IL-2, were also used as vaccine adjuvants. The results of this trial demonstrated that DNA vaccination with a PSA-coding plasmid vector, given with GM-CSF and IL-2, is safe and can induce cellular and humoural immune responses against PSA. However, a dose–response was observed with regard to induction of a PSA-specific immune response. Interestingly, in this trial, two patients that developed cellular immune response to PSA exhibited stabilisation of disease. While only one of six that did not develop PSA-reactivity showed clinical stabilisation.
Mincheff et al.[14] performed phase I/II trials to determine the safety of the PSMA vaccine after repeated i.d. injections. Twenty-six patients with PCa were entered into this toxicity-dose escalation study. Immunisations were performed i.d. at weekly intervals. Doses of DNA between 100 and 800 μg and of recombinant virus at 5×108 PFUs per application were used. They observed no immediate or long-term side effects following immunisations. All patients who received initial inoculation with the viral vector followed by PSMA plasmid boosts showed the development of a delayed-type hypersensitivity reaction after the PSMA plasmid injection. In contrast, to the patients who received a PSMA plasmid and CD86 plasmid, only 50% showed signs of successful immunisation. Of the patients who received PSMA plasmid and soluble GM-CSF, 67% were immunised. However, all patients who received the PSMA/CD86 plasmid and sGM-CSF became immunised. The patients who did not immunise during the first round were later successfully immunised after a boost with the viral vector. Further to this study, Todorova et al.[58] characterised the humoural immune response against PSMA in PCa patients. They demonstrated that PSMA is a target for humoural immune response induced by gene-based PSMA vaccination. It is also proposed that detection of anti-PSMA antibodies by immunoblot analysis and by indirect immunofluorescence could be used to monitor the vaccination effects. These results were quite encouraging, proving evidence of immune activation with different vaccination regimens. However, the heterogeneity of the medical status and the presence of concomitant hormone therapy do not permit unequivocal interpretation of the data with respect to the effectiveness of the therapy. However, several responders, as evidenced by a change in the local disease, distant metastases, and PSA levels, were identified in this cohort.
Low et al.[56] evaluated the use of EP to deliver a novel DNA vaccine (p.DOM-PSMA(27)). This vaccine encodes a domain of fragment C of tetanus toxin to induce CD4+ T cell help, fused to a tumour-derived epitope from PSMA for use in HLA-A2+ patients with recurrent PCa. In this open label phase I/II, two-arm, dose escalation trial, the DNA vaccine was delivered either by i.m injection or by i.m. injection followed by EP. Three vaccinations were given at 0, 4, and 8 weeks, with booster doses at 24 and 48 weeks. In 20 patients with first two dose cohorts, EP did not appear to add toxicity to the vaccination apart from brief and acceptable pain at injection site. They also observed highest level of humoural responses with DNA+EP strategy and these responses persisted to 18 months of follow-up. These data favour EP as a potent method for stimulating humoural responses induced by DNA vaccination in humans.
McNeel et al.[57] conducted a phase I/IIa trial with a DNA vaccine encoding human PAP in patients with stage D0 PCa. Twenty-two patients were treated in a dose-escalation trial with 100 μg, 500 μg, or 1,500 μg plasmid DNA, co-administered i.d. with 200 μg GMC-SF as a vaccine adjuvant, six times at 14-day intervals. All of these patients were observed for one year after the treatment. They did not observe any significant adverse events. Three of 22 patients developed PAP-specific IFN-γ secreting CD8+ T-cells immediately after the treatment course. While nine (41%) out of 22 patients, developed PAP-specific CD4+ and/or CD8+ T-cell proliferation. However, no humoural response (antibodies against PAP) was detected. Overall, the PSA doubling time was observed to increase from a median 6.5 months pre-treatment to 8.5 months on-treatment (P = 0.033), and 9.3 months in the 1-year post-treatment period (P = 0.054). This study provided 12 months follow up data and demonstrated that the DNA vaccine is safe, elicits an antigen-specific T-cell response, and may be associated with an increased PSA doubling time.
Conclusions and future directions
DNA vaccination for PCa is at a crucial developmental stage. The ultimate goal of any given immunotherapy including DNA vaccination is eradication of each and every cancer cell from the patient. However, this goal may be hard to achieve. DNA vaccination is a step forward in achieving immune eradication of PCa. Induction of tumour-specific T cell activation has been demonstrated with PCa DNA vaccination in both preclinical and clinical trials. However, in clinical settings, limited success has been seen in terms of tumour regression and survival. This vaccine failure may be attributed to several potential tumour escape mechanisms such as defects in antigen presentation, production of immunosuppressive substances, T cell dysfunction, and the presence of regulatory T cells [59–61]. Additionally, there is paucity in the literature of advanced clinical trials to evaluate the role of DNA vaccination in PCa. Furthermore, in the available trials there is currently a lack of long-term follow up. Ideally, the availability of data from randomised clinical trials featuring robust end points such as biochemical response, progression free and overall survival will provide categorical evidence for DNA vaccination’s potential.
In order to provoke an immune response, a tumour vaccine should not only maximise antigen-specific signals, but should also provide the necessary “co-stimulatory” environment. One approach is to include lymphokines (GM-CSF, IL-12, IL-15) or include tumour cell expression of membrane bound molecules (CD80, CD86) [62]. Furthermore, doses and schedules need optimisation; however it is clear that the immune responses depend on a primary vaccination followed by booster vaccination(s). Some studies have suggested that the best strategy for achieving an intense immune response can be priming with naked DNA followed by boosting with a viral vector [14, 63]. The use of viral vectors can enhance the immunogenicity of the vaccine due to the adjuvant properties of some of the viral products. However, naked DNA immunisation offers several potential advantages over viral mediated transduction. Among these are the inexpensive production and the inherent safety of plasmid vectors, as well as the lack of immune responses against the carrier [63].
Although the current evidence suggest that the DNA vaccines can induce immune activation resulting in PCa control, certain areas still need to be explored such as selecting the ideal antigen, identifying suitable stages of PCa for vaccine therapy, optimum dosage/schedule and adjuvant agents. Evidence suggests that it is unlikely for a single therapy to achieve the goal of curing PCa especially patients with CRPC. However, better understanding of immunobiology of PCa will lead to pathways for development of novel therapies. The perceived difficulty in translating the effectiveness of DNA vaccination from small animals to human subjects appears to have been overcome by use of EP [42]. Current data are from early stage studies and further research in this field is essential to determine the place of DNA vaccination as an adjuvant to surgery, radiotherapy and chemotherapy.
References
Center MM, Jemal A, Lortet-Tieulent J, Ward E, Ferlay J, Brawley O, Bray F: International variation in prostate cancer incidence and mortality rates. Eur Urol. 2012, 61: 1079-1092. 10.1016/j.eururo.2012.02.054.
Miller AM, Pisa P: Tumor escape mechanisms in prostate cancer. Cancer Immunol Immunother. 2007, 56: 81-87.
Schurko B, Oh WK: Docetaxel chemotherapy remains the standard of care in castration-resistant prostate cancer.Nat Clin Pract Oncol. 2008, 5: 506-507.
Coffey DS, Isaacs JT: Control of prostate growth. Urology. 1981, 17: 17-24.
Harada M, Noguchi M, Itoh K: Target molecules in specific immunotherapy against prostate cancer. Int J Clin Oncol. 2003, 8: 193-199. 10.1007/s10147-003-0332-x.
Wright GL, Grob BM, Haley C, Grossman K, Newhall K, Petrylak D, Troyer J, Konchuba A, Schellhammer PF, Moriarty R: Upregulation of prostate-specific membrane antigen after androgen-deprivation therapy. Urology. 1996, 48: 326-334. 10.1016/S0090-4295(96)00184-7.
Kawakami M, Nakayama J: Enhanced expression of prostate-specific membrane antigen gene in prostate cancer as revealed by in situ hybridization. Cancer Res. 1997, 57: 2321-2324.
McNeel DG, Nguyen LD, Storer BE, Vessella R, Lange PH, Disis ML: Antibody immunity to prostate cancer associated antigens can be detected in the serum of patients with prostate cancer. J Urol. 2000, 164: 1825-1829. 10.1016/S0022-5347(05)67114-5.
Simons JW, Mikhak B, Chang JF, DeMarzo AM, Carducci MA, Lim M, Weber CE, Baccala AA, Goemann MA, Clift SM, et al: Induction of immunity to prostate cancer antigens: results of a clinical trial of vaccination with irradiated autologous prostate tumor cells engineered to secrete granulocyte-macrophage colony-stimulating factor using ex vivo gene transfer. Cancer Res. 1999, 59: 5160-5168.
McElrath MJ: Selection of potent immunological adjuvants for vaccine construction. Semin Cancer Biol. 1995, 6: 375-385. 10.1016/1044-579X(95)90007-1.
Yedavelli SP, Guo L, Daou ME, Srivastava PK, Mittelman A, Tiwari RK: Preventive and therapeutic effect of tumor derived heat shock protein, gp96, in an experimental prostate cancer model. Int J Mol Med. 1999, 4: 243-248.
Eder JP, Kantoff PW, Roper K, Xu GX, Bubley GJ, Boyden J, Gritz L, Mazzara G, Oh WK, Arlen P, et al: A phase I trial of a recombinant vaccinia virus expressing prostate-specific antigen in advanced prostate cancer. Clin Cancer Res. 2000, 6: 1632-1638.
Small EJ, Fratesi P, Reese DM, Strang G, Laus R, Peshwa MV, Valone FH: Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J Clin Oncol. 2000, 18: 3894-3903.
Mincheff M, Tchakarov S, Zoubak S, Loukinov D, Botev C, Altankova I, Georgiev G, Petrov S, Meryman HT: Naked DNA and adenoviral immunizations for immunotherapy of prostate cancer: a phase I/II clinical trial. Eur Urol. 2000, 38: 208-217. 10.1159/000020281.
Pavlenko M, Roos AK, Lundqvist A, Palmborg A, Miller AM, Ozenci V, Bergman B, Egevad L, Hellstrom M, Kiessling R, et al: A phase I trial of DNA vaccination with a plasmid expressing prostate-specific antigen in patients with hormone-refractory prostate cancer. Br J Cancer. 2004, 91: 688-694.
Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, et al: Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010, 363: 411-422. 10.1056/NEJMoa1001294.
Rice J, Ottensmeier CH, Stevenson FK: DNA vaccines: precision tools for activating effective immunity against cancer. Nat Rev Cancer. 2008, 8: 108-120. 10.1038/nrc2326.
Stevenson FK, Ottensmeier CH, Rice J: DNA vaccines against cancer come of age. Curr Opin Immunol. 2010, 22: 264-270. 10.1016/j.coi.2010.01.019.
Joniau S, Abrahamsson PA, Bellmunt J, Figdor C, Hamdy F, Verhagen P, Vogelzang NJ, Wirth M, Van Poppel H, Osanto S: Current vaccination strategies for prostate cancer. Eur Urol. 2012, 61 (2): 290-306. 10.1016/j.eururo.2011.09.020.
Ingolotti M, Kawalekar O, Shedlock DJ, Muthumani K, Weiner DB: DNA vaccines for targeting bacterial infections. Expert Rev Vaccines. 2010, 9: 747-763. 10.1586/erv.10.57.
Bowne WB, Srinivasan R, Wolchok JD, Hawkins WG, Blachere NE, Dyall R, Lewis JJ, Houghton AN: Coupling and uncoupling of tumor immunity and autoimmunity. J Exp Med. 1999, 190: 1717-1722. 10.1084/jem.190.11.1717.
Finn OJ: Cancer vaccines: between the idea and the reality. Nat Rev Immunol. 2003, 3: 630-641. 10.1038/nri1150.
Hawkins WG, Gold JS, Dyall R, Wolchok JD, Hoos A, Bowne WB, Srinivasan R, Houghton AN, Lewis JJ: Immunization with DNA coding for gp100 results in CD4 T-cell independent antitumor immunity. Surgery. 2000, 128: 273-280. 10.1067/msy.2000.107421.
Kim JJ, Trivedi NN, Wilson DM, Mahalingam S, Morrison L, Tsai A, Chattergoon MA, Dang K, Patel M, Ahn L, et al: Molecular and immunological analysis of genetic prostate specific antigen (PSA) vaccine. Oncogene. 1998, 17: 3125-3135. 10.1038/sj.onc.1201736.
Kim JJ, Yang JS, Dang K, Manson KH, Weiner DB: Engineering enhancement of immune responses to DNA-based vaccines in a prostate cancer model in rhesus macaques through the use of cytokine gene adjuvants. Clin Cancer Res. 2001, 7: 882s-889s.
Oliveira SC, Rosinha GM, de-Brito CF, Fonseca CT, Afonso RR, Costa MC, Goes AM, Rech EL, Azevedo V: Immunological properties of gene vaccines delivered by different routes. Braz J Med Biol Res. 1999, 32: 207-214.
Moingeon P: Strategies for designing vaccines eliciting Th1 responses in humans. J Biotechnol. 2002, 98: 189-198. 10.1016/S0168-1656(02)00131-1.
Nishimura T, Nakui M, Sato M, Iwakabe K, Kitamura H, Sekimoto M, Ohta A, Koda T, Nishimura S: The critical role of Th1-dominant immunity in tumor immunology. Cancer Chemother Pharmacol. 2000, 46 (Suppl): S52-S61.
Gurunathan S, Klinman DM, Seder RA: DNA vaccines: immunology, application, and optimization*. Annu Rev Immunol. 2000, 18: 927-974. 10.1146/annurev.immunol.18.1.927.
Kongkasuriyachai D, Bartels-Andrews L, Stowers A, Collins WE, Sullivan J, Sattabongkot J, Torii M, Tsuboi T, Kumar N: Potent immunogenicity of DNA vaccines encoding Plasmodium vivax transmission-blocking vaccine candidates Pvs25 and Pvs28-evaluation of homologous and heterologous antigen-delivery prime-boost strategy. Vaccine. 2004, 22: 3205-3213. 10.1016/j.vaccine.2003.11.060.
Grossfeld GD, Stier DM, Flanders SC, Henning JM, Schonfeld W, Warolin K, Carroll PR: Use of second treatment following definitive local therapy for prostate cancer: data from the caPSURE database. J Urol. 1998, 160: 1398-1404. 10.1016/S0022-5347(01)62548-5.
MacGregor RR, Boyer JD, Ugen KE, Lacy KE, Gluckman SJ, Bagarazzi ML, Chattergoon MA, Baine Y, Higgins TJ, Ciccarelli RB, et al: First human trial of a DNA-based vaccine for treatment of human immunodeficiency virus type 1 infection: safety and host response. J Infect Dis. 1998, 178: 92-100. 10.1086/515613.
Moore AC, Hill AV: Progress in DNA-based heterologous prime-boost immunization strategies for malaria. Immunol Rev. 2004, 199: 126-143. 10.1111/j.0105-2896.2004.00138.x.
Marshall DJ, San Mateo LR, Rudnick KA, McCarthy SG, Harris MC, McCauley C, Schantz A, Geng D, Cawood P, Snyder LA: Induction of Th1-type immunity and tumor protection with a prostate-specific antigen DNA vaccine. Cancer Immunol Immunother. 2005, 54: 1082-1094. 10.1007/s00262-005-0687-0.
Trikha M, Zhou Z, Nemeth JA, Chen Q, Sharp C, Emmell E, Giles-Komar J, Nakada MT: CNTO 95, a fully human monoclonal antibody that inhibits alphav integrins, has antitumor and antiangiogenic activity in vivo. Int J Cancer. 2004, 110: 326-335. 10.1002/ijc.20116.
Mosmann TR, Coffman RL: TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989, 7: 145-173. 10.1146/annurev.iy.07.040189.001045.
Tatsumi T, Kierstead LS, Ranieri E, Gesualdo L, Schena FP, Finke JH, Bukowski RM, Mueller-Berghaus J, Kirkwood JM, Kwok WW, Storkus WJ: Disease-associated bias in T helper type 1 (Th1)/Th2 CD4(+) T cell responses against MAGE-6 in HLA-DRB10401(+) patients with renal cell carcinoma or melanoma. J Exp Med. 2002, 196: 619-628. 10.1084/jem.20012142.
Tanaka M, Kaneda Y, Fujii S, Yamano T, Hashimoto K, Huang SK, Hoon DS: Induction of a systemic immune response by a polyvalent melanoma-associated antigen DNA vaccine for prevention and treatment of malignant melanoma. Mol Ther. 2002, 5: 291-299. 10.1006/mthe.2002.0537.
Schirmbeck R, Reimann J: Revealing the potential of DNA-based vaccination: lessons learned from the hepatitis B virus surface antigen. Biol Chem. 2001, 382: 543-552.
Tangney M, Ahmad S, Collins SA, O’Sullivan GC: Gene therapy for prostate cancer. Postgrad Med. 2010, 122: 166-180. 10.3810/pgm.2010.05.2154.
Kusumanto YH, Mulder NH, Dam WA, Losen M, De Baets MH, Meijer C, Hospers GA: Improvement of in vivo transfer of plasmid DNA in muscle: comparison of electroporation versus ultrasound. Drug Deliv. 2007, 14: 273-277. 10.1080/10717540601098807.
Ahlen G, Soderholm J, Tjelle T, Kjeken R, Frelin L, Hoglund U, Blomberg P, Fons M, Mathiesen I, Sallberg M: In vivo electroporation enhances the immunogenicity of hepatitis C virus nonstructural 3/4A DNA by increased local DNA uptake, protein expression, inflammation, and infiltration of CD3+ T cells. J Immunol. 2007, 179: 4741-4753.
Dupuis M, Denis-Mize K, Woo C, Goldbeck C, Selby MJ, Chen M, Otten GR, Ulmer JB, Donnelly JJ, Ott G, McDonald DM: Distribution of DNA vaccines determines their immunogenicity after intramuscular injection in mice. J Immunol. 2000, 165: 2850-2858.
Widera G, Austin M, Rabussay D, Goldbeck C, Barnett SW, Chen M, Leung L, Otten GR, Thudium K, Selby MJ, Ulmer JB: Increased DNA vaccine delivery and immunogenicity by electroporation in vivo. J Immunol. 2000, 164: 4635-4640.
Roos AK, Pavlenko M, Charo J, Egevad L, Pisa P: Induction of PSA-specific CTLs and anti-tumor immunity by a genetic prostate cancer vaccine. Prostate. 2005, 62: 217-223. 10.1002/pros.20135.
Kuratsukuri K, Wang CY, Sone T, Nishisaka N, Jones RF, Haas GP: Induction of antibodies against prostate-specific membrane antigen (PSMA) by vaccination with a PSMA DNA vector. Eur Urol. 2002, 42: 67-73. 10.1016/S0302-2838(02)00211-7.
Kuratsukuri K, Sone T, Wang CY, Nishisaka N, Jones RF, Haas GP: Inhibition of prostate-specific membrane antigen (PSMA)-positive tumor growth by vaccination with either full-length or the C-terminal end of PSMA. Int J Cancer. 2002, 102: 244-249. 10.1002/ijc.10700.
Johnson LE, Frye TP, Chinnasamy N, Chinnasamy D, McNeel DG: Plasmid DNA vaccine encoding prostatic acid phosphatase is effective in eliciting autologous antigen-specific CD8+ T cells. Cancer Immunol Immunother. 2007, 56: 885-895. 10.1007/s00262-006-0241-8.
Johnson LE, Frye TP, Arnot AR, Marquette C, Couture LA, Gendron-Fitzpatrick A, McNeel DG: Safety and immunological efficacy of a prostate cancer plasmid DNA vaccine encoding prostatic acid phosphatase (PAP). Vaccine. 2006, 24: 293-303. 10.1016/j.vaccine.2005.07.074.
Ahmad S, Casey G, Sweeney P, Tangney M, O’Sullivan GC: Optimised electroporation mediated DNA vaccination for treatment of prostate cancer. Genet Vaccines Ther. 2010, 8: 1-10.1186/1479-0556-8-1.
Ahmad S, Casey G, Sweeney P, Tangney M, O’Sullivan GC: Prostate stem cell antigen DNA vaccination breaks tolerance to self-antigen and inhibits prostate cancer growth. Mol Ther. 2009, 17: 1101-1108. 10.1038/mt.2009.66.
Ahmad S, Casey G, Cronin M, Rajendran S, Sweeney P, Tangney M, O’Sullivan GC: Induction of effective antitumor response after mucosal bacterial vector mediated DNA vaccination with endogenous prostate cancer specific antigen. J Urol. 2011, 186: 687-693. 10.1016/j.juro.2011.03.139.
Mincheff M, Zoubak S, Makogonenko Y: Immune responses against PSMA after gene-based vaccination for immunotherapy-A: results from immunizations in animals. Cancer Gene Ther. 2006, 13: 436-444. 10.1038/sj.cgt.7700914.
Roos AK, King A, Pisa P: DNA vaccination for prostate cancer. Methods Mol Biol. 2008, 423: 463-472. 10.1007/978-1-59745-194-9_36.
Alam S, McNeel DG: DNA vaccines for the treatment of prostate cancer. Expert Rev Vaccines. 2010, 9: 731-745. 10.1586/erv.10.64.
Todorova K, Ignatova I, Tchakarov S, Altankova I, Zoubak S, Kyurkchiev S, Mincheff M: Humoral immune response in prostate cancer patients after immunization with gene-based vaccines that encode for a protein that is proteasomally degraded. Cancer Immun. 2005, 5: 1-
Low L, Mander A, McCann K, Dearnaley D, Tjelle T, Mathiesen I, Stevenson F, Ottensmeier CH: DNA vaccination with electroporation induces increased antibody responses in patients with prostate cancer. Hum Gene Ther. 2009, 20: 1269-1278. 10.1089/hum.2009.067.
McNeel DG, Dunphy EJ, Davies JG, Frye TP, Johnson LE, Staab MJ, Horvath DL, Straus J, Alberti D, Marnocha R, et al: Safety and immunological efficacy of a DNA vaccine encoding prostatic acid phosphatase in patients with stage D0 prostate cancer. J Clin Oncol. 2009, 27: 4047-4054. 10.1200/JCO.2008.19.9968.
Kiessling R, Wasserman K, Horiguchi S, Kono K, Sjoberg J, Pisa P, Petersson M: Tumor-induced immune dysfunction. Cancer Immunol Immunother. 1999, 48: 353-362. 10.1007/s002620050586.
Pawelec G: Tumour escape: antitumour effectors too much of a good thing?. Cancer Immunol Immunother. 2004, 53: 262-274. 10.1007/s00262-003-0469-5.
Ahmad M, Rees RC, Ali SA: Escape from immunotherapy: possible mechanisms that influence tumor regression/progression. Cancer Immunol Immunother. 2004, 53: 844-854.
Tangney M, Casey G, Larkin JO, Collins CG, Soden D, Cashman J, Whelan MC, O’Sullivan GC: Non-viral in vivo immune gene therapy of cancer: combined strategies for treatment of systemic disease. Cancer Immunol Immunother. 2006, 55: 1443-1450. 10.1007/s00262-006-0169-z.
Mincheff M, Altankova I, Zoubak S, Tchakarov S, Botev C, Petrov S, Krusteva E, Kurteva G, Kurtev P, Dimitrov V, et al: In vivo transfection and/or cross-priming of dendritic cells following DNA and adenoviral immunizations for immunotherapy of cancer–changes in peripheral mononuclear subsets and intracellular IL-4 and IFN-gamma lymphokine profile. Crit Rev Oncol Hematol. 2001, 39: 125-132. 10.1016/S1040-8428(01)00111-1.
Acknowledgement
This work was supported by the Cork Cancer Research Centre.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SA carried out literature search, interpreted the data and drafted the manuscript. MT, PS and GCOS helped with data interpretation and drafting the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ahmad, S., Sweeney, P., Sullivan, G.C. et al. DNA vaccination for prostate cancer, from preclinical to clinical trials - where we stand?. Genet Vaccines Ther 10, 9 (2012). https://doi.org/10.1186/1479-0556-10-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1479-0556-10-9