
Abstract
Recent studies of the primary cilium have begun to provide further insights into ciliary ultrastructure, with an emerging picture of complex compartmentalization and molecular components that combine in functional modules. Many proteins that are mutated in ciliopathies are localized to the transition zone, a compartment of the proximal region of the cilium. The loss of these components can disrupt ciliary functions such as the control of protein entry and exit from the cilium, the possible trafficking of essential ciliary components, and the regulation of signaling cascades and control of the cell cycle. The discovery of functional modules within the primary cilium may help in understanding the variable phenotypes and pleiotropy in ciliopathies.
Similar content being viewed by others

Review
Cilia are microtubule-based, hair-like organelles that occur on the apical surface of most mammalian cells in G0/G1 of the cell cycle [1], with the exception of bone marrow-derived cells [2]. Defects in cilia structure or function are the cause of a suite of congenital conditions known as ciliopathies, which now include polycystic kidney disease, nephronophthisis, Senior-Løken syndrome, Bardet-Biedl syndrome (BBS), Joubert syndrome (JBTS) and Meckel-Gruber syndrome (MKS). Most of these conditions vary in the severity of the clinical phenotype, and display extensive allelism [3] and pleiotropy [3–7].
Ultrastructure of the cilium
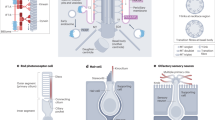
There are three general types of cilia, differentiated on the basis of their microtubule structure (Figure 1). Cilia with the canonical “9 + 2”microtubule pattern are motile: Motile cilia occur on epithelial cells in the lungs, in the olfactory bulb, in the inner ear as a kinocilium, and in the reproductive tracts of both sexes. Motile cilia in the respiratory tract perform “whip”-like movements to mediate fluid flow [8], although olfactory cilia, despite their 9 + 2 microtubule pattern, are thought to be immotile [9] and kinocilia are moved by deflection [10]. During embryonic development nodal cilia play a crucial role in left-right patterning. They are located at the embryonic node, and although they have a “9 + 0” microtubule pattern they mediate a leftward flow at the node in a “whirlpool”-like manner due to the retention of dynein arms between microtubules. The leftward flow is thought to transport morphogens that are essential for the first step of symmetry breaking in the developmental of the mammalian body plan [2]. The third type of cilia is an immotile (“9 + 0”) primary cilium, which is now known to participate in diverse roles in cell signaling, chemosensation, mechanosensation and thermosensation [11].

This figure shows schematic representation of cilia ultrastructure with cross-sections at the level of the basal body, transition fibers, transition zone and axoneme in motile and non-motile cilia. On the left hand side, is an enlarged immunofluorescence micrograph of a single primary cilium on an IMCD3 epithelial cell immunostained for the cilia marker acetylated α-tubulin (blue), the basal body marker γ-tubulin (red), and transmembrane protein (TMEM) 216 (green) which localizes to the transition zone. Scale bar = 1 μm.
In quiescent cells, cilia are derived from a mother centriole that migrates to the apical surface of the cell and matures into the basal body. The mother centriole consists of a barrel-shaped structure of nine triplets of microtubules, each triplet built from A-, B- and C-tubules (Figure 1). The basal body consists of both mother and daughter centrioles, but only the former acts as a matrix for the subsequent nucleation of microtubules during the formation of a cilium. The mother centriole can also be distinguished from the daughter by the presence of fibrous distal and subdistal appendages [12]. During ciliogenesis, the doublets containing A- and B-tubules are extended to form the ciliary axoneme by a process of intraflagellar transport (IFT) [13] and the centriolar appendages mature into transition fibers [14]. Once the basal body is docked, the axoneme begins to be assembled. The microtubules mediate anterograde transport towards the tip of cilia by kinesin motors carrying IFT-complex B proteins and other cargo proteins. In turn, cytoplasmic dyneins mediate retrograde movement of IFT-complex A towards the proximal regions of the cilium. IFT transport plays a crucial role in cilia assembly and disassembly as well as in transporting signaling components. Dyneins are also components of more general motor protein complexes responsible for minus-end, microtubule-based motile processes. IFT mediates both assembly and resorbtion of the cilium, and processing of key intermediates of signaling cascades [15].
The transition zone
The most proximal region of a cilium is called the “ciliary gate” and can be divided into two structurally distinct sub-regions: the transition fibers and the transition zone (Figure 1) [16]. For many years, the ciliary gate was observed in TEM (transmission electron microscopy) cross-sections [16–19] but its function remained unknown. These early observations suggest that the ciliary gate forms at the very early stages of ciliogenesis that precede IFT. The basal body terminates with the end of the C-tubule and the beginning of the transition fibers. More distally, the axoneme of the cilium is then established. In motile cilia, the boundary between the axoneme and the transition zone is known as the basal plate and is thought to take part in the nucleation of the central microtubules (Figure 1) [20]. The appearance of the transition zone varies between species and cell-type [21], but the basic structural components appear to be conserved. For example, the so-called “connecting cilium” in photoreceptor cells of mammals is a ciliary transition zone that extends between the outer and inner segment of photoreceptors [22], and its extended structure was a factor in enabling the biochemical purification of outer segments and the identification of the photoreceptor sensory cilium proteome [23].
Transition fibers (Figure 1) emerge from the B-tubules of the basal body triplet microtubules just before the end of the C-tubule and form a “pinwheel-like” structure on TEM cross-sections. The tips of transition fibers are thought to anchor microtubules to the plasma membrane, although the composition of transition fibers is still largely unknown. In some species and in motile cilia the terminal plate (often visualized as an electron-dense aggregate on TEM) is observed in this area. No function has been ascribed for the terminal plate, although in Tetrahymena it was shown to be enriched in centrin [24]. Transition fibers are observed on the mature mother centriole and they may play a role in anchoring to the plasma membrane through CEP164 [25] and ODF2 (outer dense fibre 2)/cenexin [26]. In Chlamydomonas, IFT52 was observed on transition fibers as well [27], suggesting that they have a role in docking the IFT and motor proteins required for ciliogenesis. Although the complete protein composition of those fibers is still unknown, it is thought that they take part in creating a pore complex, similar to nuclear pores, which is required for transporting proteins in and out of cilia [28].
Moving up toward the ciliary tip and on the other side of the ciliary gate, the so-called “Y-shaped” linkers and the ciliary necklace are observed (Figure 1), the latter a characteristic structure of the transition zone. Y-shaped linkers are structures connecting the outer doublets of microtubules to the plasma membrane and the ciliary necklace [29]. Their detailed protein structure is as yet uncharacterized and their shape and name is species specific [20, 29–34]. The ciliary necklace is a specialized structure that consists of several parallel strands of intramembrane particles and their number is species and cell-specific. The identity of these strands is unknown, but they encircle the ciliary membrane spacing from the plasma/ciliary membrane boundary to the basal plate [20]. Y-shaped linkers and the ciliary necklace are especially visible in the elongated transition zone structure of connecting cilia in photoreceptors [35], and the latter has been shown to contain CEP290 in Chlamydomonas[36] and RPGRIP1L in C.elegans[19]. In motile cilia, the ciliary necklace coincides with the minus ends of the central pair of microtubules at the basal plate [20]. This region is enriched in γ-tubulin [37], which suggests that this is the location for nucleation and stabilisation of the central pair of microtubules. Similar to the transition fibers, the transition zone has been proposed to regu-late ciliary protein composition in Chlamydomonas, C. elegans and mammalian cells [19, 36, 38] by regulating intracellular trafficking to and from the cilium. However, the molecular details of protein sorting at the transition zone remain to be discovered.
Trafficking to cilia
It is difficult to define a simple model for the mechanisms of protein targeting to cilia, with several competing models described below and reviewed elsewhere [39–41]. Although the identity of ciliary cargo proteins is becoming clearer, the mechanisms of their intracellular transport are still mysterious. One model assumes the existence of characteristic protein recognition sequences that mediate correct targeting. A focus of research has been the ciliary transport of transmembrane proteins, which has required the identification of specific protein sequences necessary for targeting and the identification of modules that recognize these sequences [42–44]. These include, for example, the N-terminal RVxP sequence for the PKD2 protein [44], and the C- terminal VxPx sequence for rhodopsin [43].
A second model involves vesicle trafficking and exocytosis, although the direct vesicle transport of membrane proteins through the ciliary gate is unlikely because the vesicle size exceeds 60 nm. Instead, proteins are thought to be transported in vesicles from the Golgi apparatus to a specific docking site at the periciliary base. The exocyst complex is then thought to tether the vesicles, presumably directing the fusion of vesicles with the ciliary membrane mediated by SNAREs (soluble N- ethylmaleimide sensitive factor receptors) and the Rab family of small GTPases [45]. SNAREs are present at the surface of the exocyst complex to allow proteins to pass the ciliary diffusion barrier and to cross to the ciliary membrane [45], implying an active transport process [41]. This model of trafficking would therefore be analogous to that proposed for nuclear pores where importins and exportins are utilized. IFT proteins may mediate this transport machinery since they are enriched at the level of transition fibers [27]. Once trafficked through the ciliary gate, proteins may then be incorporated as cargo proteins in the IFT complexes and transported along the ciliary axoneme. The third model suggests that IFT proteins create clusters in the trans-Golgi network that coat vesicles and ensure targeted trafficking to the cilium [46]. The clusters would later become IFT complexes still attached to cargo proteins.
However, studies of Rab8 seem to confirm the vesicle theory, and clearly suggest that Rab GTPases coordinate with each other in the regulation of vesicular trafficking during primary ciliogenesis. Rab8 is a member of the Rab family of small GTPases that regulates membrane traffic from endosomal compartments to the cell surface [47] as well as mediating transport, docking and fusion of vesicles with acceptor modules at the base of the primary cilium [48]. Rab8 plays an essential role in ciliogenesis [49–51] and acts downstream of Rab11 and Rabin8, the guanine nucleotide exchange factor (GEF) that activates Rab8 [52]. An additional downstream binding partner of Rab8 is CEP290, which has been shown to localize to the pericentrosomal compartment, basal body and transition zone [53, 54]. CEP290 plays an important role at the ciliary gate, where together with other proteins containing C2/B9-related domains predicted to bind phospholipids [55], it is proposed to be involved in membrane/vesicle trafficking and fusion [53]. In particular, CC2D2A, a C2 domain-containing protein, is now also suggested to mediate vesicle trafficking and fusion since it localizes to the photoreceptor connecting cilium/transition zone, and appears to facilitate protein transport through Rab8-dependent processes [56].
Another possible mechanism of protein targeting to cilia is through the BBSome complex. This is proposed to mediate trafficking of transmembrane proteins to the ciliary membrane and consists of seven highly conserved proteins, one novel protein and a coat-like structure [50]. The BBSome directly recognizes cilia targeting sequences and is the major effector of Arl6/BBS3 (an Arf-like GTPase). Rabin8, the GEF that activates Rab8, has also been shown to interact with the BBSome [50]. The BBSome is not directly required for cilia formation in most tissues [57] but its failure to deliver important receptors and transmembrane proteins to the cilium is thought to result in cell signaling failure and organ-specific pathological abnormalities. Proteins, after fulfilling their function in the cilium, have to be removed from it. This function is suggested to be also mediated by the BBSome. In Chlamydomonas it was observed that the BBSome and IFT particles co-localize, and the BBSome may play a docking function for the retrograde IFT complex A [58]. Proteins targeted to be removed from cilia may also be ubiquitinated, then accumulated at the base of cilia for targeted degradation by the endocytic pathway [59].
Recent advances in characterizing the transition zone
In five recent papers, the composition of protein complexes at the ciliary gate and transition zone was investigated. In two of the studies [54, 60], the authors used the G-LAP tandem-affinity method, a stringent proteomic strategy for the identification of interacting proteins. It allowed the authors not only to identify three interconnected functional modules, but to also identify three new ciliopathy genes: Sang et al. 2011 identified ATXN10 and TCTN2, and Garcia-Gonzalo et al. 2011 identified TCTN1. The first module, the “nephronophthisis NPHP module” identified in Sang et al. 2011, was composed of NPHP1/nephrocystin-1, which was previously suggested to take part in the regulation of cargo and IFT ciliary entry [61]. NPHP4 and RPGRIP1L (also components of the NPHP module) were also localized to the transition zone and cell-cell boundaries. In vitro studies showed that this module was not essential for ciliogenesis, but it may play an important part in epithelial morphogenesis and the establishment of tissue architecture. On the other hand, a second module (the “JBTS module”) that contains IQCB1 and CEP290 is essential for cilia formation. Proteins in the third module (the “MKS module” containing MKS1, CC2D2A and TCTN2) are involved in neural tube development and Sonic Hedgehog signaling. The authors proposed the existence of an additional ciliary segment located distal to the transition zone in the proximal part of cilia, which they called the “inversin compartment” (Figure 2). The function of this section of the cilium remains to be understood, but several proteins were found to be localized in it, notably NEK8, INVS/inversin and NPHP3 [62]. INVS and AHI1/jouberin were found to play a bridging role between these three modules. There were no interactions observed between the NPHP-JBTS-MKS modules and the BBS proteins, and no overlap with IFT complex proteins [60].

Localization of the indicated proteins (coloured ovals) to sub-ciliary compartments (pale blue, axoneme; grey, inversin compartment; pink, transition zone; dark blue, basal body; grey with dashed square , centriolar satellites) are represented based on biochemical and genetic interaction data from four recent studies of the composition of the transition zone (Sang et al. 2011, Garcia- Gonzalo et al. 2011, Williams et al. 2011 and Huang et al. 2011; each indicated at the top of each section). White text indicates common components identified by different studies; dark lines indicate an interaction identified by MS/MS; a dashed line indicates a genetic interaction; overlapping ellipses indicated a direct interaction. The model system(s) used to generate these data are also indicated for each study.
In Garcia-Gonzalo et al. 2011, the authors identified mutations in TCTN1 as a cause of Joubert syndrome. They showed that TCTN1/tectonic-1 interacts with TCTN2, TCTN3, MKS1, B9D1 and CC2D2A and, under some circumstances, with CEP290, TMEM67/meckelin and TMEM216. They showed that TCTN1 localizes to the transition zone and that it is required for transition zone localization of MKS1 and TMEM67. During detailed studies they observed other proteins at the transition zone: TCTN2, TCTN3 and TMEM67 (all three of which also localized elsewhere in the cilium); MKS1 and CEP290 (also localized to centriolar appendages); CC2D2A and B9D1 (also localized to the basal body) and NPHP4. RPGRIP1L was localized to the basal body like TMEM216, which was also observed at distal regions of the cilium and occasionally at the transition zone (Figure 1). The authors stated that the Tectonic module (containing TCTN1, TCTN2 and TCTN3) and MKS proteins localize to the transition zone where they regulate ciliogenesis and ciliary membrane composition in a tissue-specific manner, and dysfunction of the transition zone is a basic defect causing human ciliopathies [54].
Williams and colleagues, 2011 conducted their research on C. elegans which has ciliated sensory neurons in amphid sensory cells. They grouped the investigated proteins into two modules: MKS/MKSR (containing MKS1, B9D1, B9D2, TMEM67, CC2D2A) and NPHP (containing NPHP1 and NPHP4) (Figure 2) that despite seemingly unrelated components share a common biological function. Both modules are linked through RPGRIP1L, which is responsible for endogenous localization to the transition zone for all of the other investigated proteins. Collectively, both modules are required for the early steps of ciliogenesis, namely the basal body – transition zone membrane docking and formation of the transition zone [19]. It is notable that CC2D2A has been implicated in Rab8-dependent vesicle trafficking and fusion [56], although the role of the MKS/MKSR module in mediating these processes remains unclear.
Li et al. 2011 [63] investigated TCTEX1/DYNLT, a dynein light chain subunit involved in cargo binding but that can also be recruited for functions independent of the dynein complex [64]. Active phosphorylated TCTEX1 was found to be recruited to the ciliary transition zone prior to cilia disassembly and entry into S phase of the cell cycle [63]. Furthermore, active TCTEX1 was recruited to the transition zone of neuronal progenitor cells, where it had a key role in cell cycle regulation and the probable determination of cell fate between proliferation of progenitors and differentiation into post-mitotic neurons.
In Huang et al. 2011, the authors identified mutations in TMEM237/JBTS14 as a cause of Joubert syndrome-related disorders [65]. They used three model systems (C. elegans, zebrafish and IMCD3 cells) to investigate biochemical and genetic interactions between TMEM237 and other ciliary proteins. TMEM237 localized to the ciliary transition zone and, largely in agreement with previous studies [54, 60], interacted genetically with NPHP4, TMEM216, B9D1 and B9D2. The authors also noted that TMEM237 required RPGRIP1L for localization to the transition zone in both the IMCD3 cell and C. elegans models, which suggests that RPGRIP1L has a structural role in mediating the scaffolding or bridging between interacting ciliary proteins [65].
Figure 2 summarizes the localization of ciliary proteins based on the results of Sang et al. 2011, Garcia-Gonzalo et al. 2011, Williams et al. 2011 and Huang et al. 2011. There are some obvious inconsistencies among the studies, which could be explained by possible differences in cilia structure between tissues and the dynamic nature of ciliogenesis and protein transport within cilia. Sang et al. 2011 proposed interaction modules that will be very useful in further analysis of protein-protein interactions, signaling pathways and the molecular structure of cilia. Garcia-Gonzalo el al. 2011 added another module containing Tectonic proteins (TCTN1, TCTN2 and TCTN3) crucial for cilia formation and the patterning of the developing embryo. Williams et al. 2011 described a further functional module (the “MKS/MKSR module” including MKS1, TMEM67, CC2D2A, B9D1 and B9D2). This observation confirmed the existence of a general “NPHP1-NPHP4-RPGRIP1L module” with RPGRIP1L functioning as a bridging protein. Huang et al. 2011 described a fourth ciliopathy geneTMEM237 using a next-generation sequencing strategy, and confirmed the transition zone localization of RPGRIP1L, NPHP4, B9D1 and B9D2. The dynamic nature of ciliogenesis regulation by transition zone proteins has been highlighted by a fourth recent study [63].
Conclusion
In conclusion, four recent studies [19, 54, 60, 65] support the concept of human ciliopathies being caused by sorting defects at the transition zone (Table 1), and the ciliary gate playing a crucial role in cilia assembly and selective regulation of cilia protein content. What remains unexplained is the function of ciliary modules in mediating cilia trafficking, how these could regulate the signaling cascades that are mediated by cilia, and the connection with other complexes such as the inversin module and the BBSome. It seems likely that the elucidation of these mechanistic details will begin to explain the phenotypic variability and pleiotropy of human ciliopathies (Figure 3). These could arise from either the diverse requirements of the protein composition of transition zones in different tissues, or the influence of modifier alleles in interacting components of individual functional modules such as TTC21B or RPGRIP1L [3, 66]. A fifth study [63] has highlighted the complex, dynamic nature of the transition zone and a possible role of this region of the cilium in G1/S checkpoint control. Linking the cilium, cell cycle control and extracellular cues of signaling pathways will be a further field of intensive future work, and will no doubt bring further surprises in our understanding of the complex ultrastructure of the primary cilium.

Five ciliopathies with extensive genetic heterogeneity and pleiotropy (BBS, Bardet-Biedl syndrome; JBTS, Joubert syndrome; MKS, Meckel-Gruber syndrome; NPHP, nephronophthisis; SLS, Senior-Løken syndrome) are indicated by ovals of different colour, with the ciliary proteins mutated as a known cause of each disorder indicated by the coloured lines.
Abbreviations
- BBS:
-
Bardet-Biedl syndrome
- IFT:
-
Intraflagellar transport
- JBTS:
-
Joubert syndrome
- LCA:
-
Leber congenital amaurosis
- MKS:
-
Meckel-Gruber syndrome
- NPHP:
-
Nephronophthisis
- TEM:
-
Transmission electron microscopy
- TMEM:
-
Transmembrane protein
- SLS:
-
Senior-Løken syndrome
- SNARE:
-
Soluble N- ethylmaleimide sensitive factor receptor
- GEF:
-
Guanine nucleotide exchange factor.
References
Pan J, Snell W: The Primary Cilium: Keeper of the Key to Cell Division. Cell. 2007, 129: 1255-1257. 10.1016/j.cell.2007.06.018.
Praetorius HA, Spring KR: A physiological view of the primary cilium. Annu Rev Physiol. 2005, 67: 515-529. 10.1146/annurev.physiol.67.040403.101353.
Davis EE, Zhang Q, Liu Q, Diplas BH, Davey LM, Hartley J, Stoetzel C, Szymanska K, Ramaswami G, Logan CV, Muzny DM, Young AC, Wheeler DA, Cruz P, Morgan M, Lewis LR, Cherukuri P, Maskeri B, Hansen NF, Mullikin JC, Blakesley RW, Bouffard GG, Comparative Sequencing Program NISC, Gyapay G, Rieger S, Tönshoff B, Kern I, Soliman NA, Neuhaus TJ, Swoboda KJ, Kayserili H: TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet. 2011, 43: 189-196. 10.1038/ng.756.
Baala L, Romano S, Khaddour R, Saunier S, Smith UM, Audollent S, Ozilou C, Faivre L, Laurent N, Foliguet B, Munnich A, Lyonnet S, Salomon R, Encha-Razavi F, Gubler MC, Boddaert N, de Lonlay P, Johnson CA, Vekemans M, Antignac C, Attie-Bitach T: The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am J Hum Genet. 2007, 80: 186-194. 10.1086/510499.
Bergmann C, Fliegauf M, Bruchle NO, Frank V, Olbrich H, Kirschner J, Schermer B, Schmedding I, Kispert A, Kranzlin B, Nürnberg G, Becker C, Grimm T, Girschick G, Lynch SA, Kelehan P, Senderek J, Neuhaus TJ, Stallmach T, Zentgraf H, Nürnberg P, Gretz N, Lo C, Lienkamp S, Schäfer T, Walz G, Benzing T, Zerres K, Omran H: Loss of nephrocystin-3 function can cause embryonic lethality, meckel-gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia. Am J Hum Genet. 2008, 82: 959-970. 10.1016/j.ajhg.2008.02.017.
Devuyst O, Arnould VJ: Mutations in RPGRIP1L: extending the clinical spectrum of ciliopathies. Nephrol Dial Transplant. 2008, 23: 1500-1503. 10.1093/ndt/gfn033.
Mougou-Zerelli S, Thomas S, Szenker E, Audollent S, Elkhartoufi N, Babarit C, Romano S, Salomon R, Amiel J, Esculpavit C, Gonzales M, Escudier E, Leheup B, Loget P, Odent S, Roume J, Gérard M, Delezoide AL, Khung S, Patrier S, Cordier MP, Bouvier R, Martinovic J, Gubler MC, Boddaert N, Munnich A, Encha-Razavi F, Valente EM, Saad A, Saunier S, et al: CC2D2A Mutations in Meckel and Joubert Syndromes Indicate a Genotype-Phenotype Correlation. Hum Mutat. 2009, 30: 1574-1582. 10.1002/humu.21116.
Kotov N, Bates D, Gizatullina A, Gilaziev B, Khairullin R, Chen M, Drozdov I, Umezawa Y, Hundhausen C, Aleksandrov A, Yan XG, Spurgeon SK, Smales CM, Valeyev NV: Computational modelling elucidates the mechanism of ciliary regulation in health and disease. BMC Systems Biology. 2011, 5: 143-10.1186/1752-0509-5-143.
Menini A (Ed:: The Neurobiology of Olfaction. Boca Raton, FL: CRC Press; 2010
Fontilla MF, Peterson EH: Kinocilia heights on utricular hair cells. Hear Res. 2000, 145: 8-16. 10.1016/S0378-5955(00)00068-X.
Fliegauf M, Benzing T, Omran H: When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007, 8: 880-893. 10.1038/nrm2278.
Paintrand M, Moudjou M, Delacroix H, Bornens M: Centrosome organization and centriole architecture: Their sensitivity to divalent cations. J Struct Biol. 1992, 108: 107-128. 10.1016/1047-8477(92)90011-X.
Satir P: Studies on cilia. J Cell Biol. 1968, 39: 77-94. 10.1083/jcb.39.1.77.
Sorokin SP: Reconstructions of Centriole Formation and Ciliogenesis in Mammalian Lungs. J Cell Sci. 1968, 3: 207-230.
Pedersen LB, Veland IR, Schroder JM, Christensen ST: Assembly of primary cilia. Dev Dyn. 2008, 237: 1993-2006. 10.1002/dvdy.21521.
Omran H: NPHP proteins: gatekeepers of the ciliary compartment. J Cell Biol. 2010, 190: 715-717. 10.1083/jcb.201008080.
Gibbons IR, Grimstone AV: On Flagellar Structure in Certain Flagellates. J Biophys Biochem Cytol. 1960, 7: 697-716. 10.1083/jcb.7.4.697.
Ringo DL: Flagellar motion and fine structure of the flagellar apparatus in Chlamydomonas. J Cell Biol. 1967, 33: 543-571. 10.1083/jcb.33.3.543.
Williams CL, Li C, Kida K, Inglis PN, Mohan S, Semenec L, Bialas NJ, Stupay RM, Chen N, Blacque OE, Yoder BK, Leroux MR: MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. The Journal of Cell Biology. 2011, 192: 1023-1041. 10.1083/jcb.201012116.
Gilula NB, Satir P: The ciliary necklace. J Cell Biol. 1972, 53: 494-509. 10.1083/jcb.53.2.494.
Fisch C, Dupuis-Williams P: Ultrastructure of cilia and flagella – back to the future!. Biol Cell. 2011, 103: 249-270. 10.1042/BC20100139.
Quinlan RJ, Tobin JL, Beales PL: Modeling ciliopathies: primary cilia in development and disease. Curr Top Dev Biol. 2008, 84: 249-310.
Liu Q, Tan G, Levenkova N, Li T, Pugh EN, Rux JJ, Speicher DW, Pierce EA: The Proteome of the Mouse Photoreceptor Sensory Cilium Complex. Mol Cell Proteomics. 2007, 6: 1299-1317. 10.1074/mcp.M700054-MCP200.
Kilburn CL, Pearson CG, Romijn EP, Meehl JB, Giddings TH, Culver BP, Yates JR, Winey M: New Tetrahymena basal body protein components identify basal body domain structure. J Cell Biol. 2007, 178: 905-912. 10.1083/jcb.200703109.
Graser S, Stierhof Y-D, Lavoie SB, Gassner OS, Lamla S, Le Clech M, Nigg EA: Cep164, a novel centriole appendage protein required for primary cilium formation. J Cell Biol. 2007, 179: 321-330. 10.1083/jcb.200707181.
Ishikawa H, Kubo A, Tsukita S, Tsukita S: Odf2-deficient mother centrioles lack distal/subdistal appendages and the ability to generate primary cilia. Nat Cell Biol. 2005, 7: 517-524. 10.1038/ncb1251.
Deane JA, Cole DG, Seeley ES, Diener DR, Rosenbaum JL: Localization of intraflagellar transport protein IFT52 identifies basal body transitional fibers as the docking site for IFT particles. Curr Biol. 2001, 11: 1586-1590. 10.1016/S0960-9822(01)00484-5.
Rosenbaum JL, Witman GB: Intraflagellar transport. Nat Rev Mol Cell Biol. 2002, 3: 813-825. 10.1038/nrm952.
O'Toole ET, Giddings TH, Dutcher SK: Understanding Microtubule Organizing Centers by Comparing Mutant and Wild‐Type Structures with Electron Tomography. Methods in Cell Biology. Edited by: McIntosh JR. 2007, Academic Press, Waltham, MS, 125-143.
Reese TS: Olfactory cilia in the frog. J Cell Biol. 1965, 25: 209-230.
Anderson RGW: Isolation of ciliated or inciliated basal bodies from the rabbit oviduct. J Cell Biol. 1974, 60: 393-404. 10.1083/jcb.60.2.393.
Cavalier-Smith T: Basal Body and Flagellar Development During the Vegetative Cell Cycle and the Sexual Cycle of Chlamydomonas Reinhardii. J Cell Sci. 1974, 16: 529-556.
Dute R, Kung C: Ultrastructure of the proximal region of somatic cilia in Paramecium tetraurelia. J Cell Biol. 1978, 78: 451-464. 10.1083/jcb.78.2.451.
Geimer S, Melkonian M: Centrin Scaffold in Chlamydomonas reinhardtii Revealed by Immunoelectron Microscopy. Eukaryot Cell. 2005, 4: 1253-1263. 10.1128/EC.4.7.1253-1263.2005.
Horst CJ, Forestner DM, Besharse JC: Cytoskeletal-membrane interactions: a stable interaction between cell surface glycoconjugates and doublet microtubules of the photoreceptor connecting cilium. J Cell Biol. 1987, 105: 2973-2987. 10.1083/jcb.105.6.2973.
Craige B, Tsao C-C, Diener DR, Hou Y, Lechtreck K-F, Rosenbaum JL, Witman GB: CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J Cell Biol. 2010, 190: 927-940. 10.1083/jcb.201006105.
Silflow CD, Liu B, LaVoie M, Richardson EA, Palevitz BA: γ-tubulin in Chlamydomonas: Characterization of the gene and localization of the gene product in cells. Cell Motil Cytoskeleton. 1999, 42: 285-297. 10.1002/(SICI)1097-0169(1999)42:4<285::AID-CM3>3.0.CO;2-Z.
Hu Q, Milenkovic L, Jin H, Scott MP, Nachury MV, Spiliotis ET, Nelson WJ: A Septin Diffusion Barrier at the Base of the Primary Cilium Maintains Ciliary Membrane Protein Distribution. Science. 2010, 329: 436-439. 10.1126/science.1191054.
Ishikawa H, Marshall WF: Ciliogenesis: building the cell's antenna. Nat Rev Mol Cell Biol. 2011, 12: 222-234. 10.1038/nrm3085.
Hu Q, Nelson WJ: Ciliary diffusion barrier: The gatekeeper for the primary cilium compartment. Cytoskeleton. 2011, 68: 313-324. 10.1002/cm.20514.
Nachury MV, Seeley ES, Jin H: Trafficking to the Ciliary Membrane: How to Get Across the Periciliary Diffusion Barrier?. Annu Rev Cell Dev Biol. 2010, 26: 59-87. 10.1146/annurev.cellbio.042308.113337.
Deretic D, Williams AH, Ransom N, Morel V, Hargrave PA, Arendt A: Rhodopsin C terminus, the site of mutations causing retinal disease, regulates trafficking by binding to ADP-ribosylation factor 4 (ARF4). Proc Natl Acad Sci U S A. 2005, 102: 3301-3306. 10.1073/pnas.0500095102.
Mazelova J, Astuto-Gribble L, Inoue H, Tam BM, Schonteich E, Prekeris R, Moritz OL, Randazzo PA, Deretic D: Ciliary targeting motif VxPx directs assembly of a trafficking module through Arf4. EMBO J. 2009, 28: 183-192. 10.1038/emboj.2008.267.
Geng L, Okuhara D, Yu Z, Tian X, Cai Y, Shibazaki S, Somlo S: Polycystin-2 traffics to cilia independently of polycystin-1 by using an N-terminal RVxP motif. J Cell Sci. 2006, 119: 1383-1395. 10.1242/jcs.02818.
Jahn R, Scheller RH: SNAREs — engines for membrane fusion. Nat Rev Mol Cell Biol. 2006, 7: 631-643. 10.1038/nrm2002.
Follit JA, Tuft RA, Fogarty KE, Pazour GJ: The Intraflagellar Transport Protein IFT20 Is Associated with the Golgi Complex and Is Required for Cilia Assembly. Mol Biol Cell. 2006, 17: 3781-3792. 10.1091/mbc.E06-02-0133.
Ang AL, Taguchi T, Francis S, Fölsch H, Murrells LJ, Pypaert M, Warren G, Mellman I: Recycling endosomes can serve as intermediates during transport from the Golgi to the plasma membrane of MDCK cells. J Cell Biol. 2004, 167: 531-543. 10.1083/jcb.200408165.
Hsiao Y-C, Tong ZJ, Westfall JE, Ault JG, Page-McCaw PS, Ferland RJ: Ahi1, whose human ortholog is mutated in Joubert syndrome, is required for Rab8a localization, ciliogenesis and vesicle trafficking. Hum Mol Genet. 2009, 18: 3926-3941. 10.1093/hmg/ddp335.
Yoshimura S-i, Egerer J, Fuchs E, Haas AK, Barr FA: Functional dissection of Rab GTPases involved in primary cilium formation. J Cell Biol. 2007, 178: 363-369. 10.1083/jcb.200703047.
Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peränen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC, Jackson PK: A Core Complex of BBS Proteins Cooperates with the GTPase Rab8 to Promote Ciliary Membrane Biogenesis. Cell. 2007, 129: 1201-1213. 10.1016/j.cell.2007.03.053.
Follit JA, Li L, Vucica Y, Pazour GJ: The cytoplasmic tail of fibrocystin contains a ciliary targeting sequence. J Cell Biol. 2010, 188: 21-28. 10.1083/jcb.200910096.
Knödler A, Feng S, Zhang J, Zhang X, Das A, Peränen J, Guo W: Coordination of Rab8 and Rab11 in primary ciliogenesis. Proc Natl Acad Sci. 2010, 107: 6346-6351. 10.1073/pnas.1002401107.
Kim J, Krishnaswami SR, Gleeson JG: CEP290 interacts with the centriolar satellite component PCM-1 and is required for Rab8 localization to the primary cilium. Hum Mol Genet. 2008, 17: 3796-3805. 10.1093/hmg/ddn277.
Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, Seol AD, Robinson JF, Bennett CL, Josifova DJ, García-Verdugo JM, Katsanis N, Hildebrandt F, Reiter JF: A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. 2011, 43: 776-784. 10.1038/ng.891.
Nalefski EA, Falke JJ: The C2 domain calcium-binding motif: Structural and functional diversity. Protein Sci. 1996, 5: 2375-2390. 10.1002/pro.5560051201.
Bachmann-Gagescu R, Phelps IG, Stearns G, Link BA, Brockerhoff SE, Moens CB, Doherty D: The ciliopathy gene cc2d2a controls zebrafish photoreceptor outer segment development through a role in Rab8-dependent vesicle trafficking. Hum Mol Genet. 2011, 20: 4041-4055. 10.1093/hmg/ddr332.
Jin H, White SR, Shida T, Schulz S, Aguiar M, Gygi SP, Bazan JF, Nachury MV: The Conserved Bardet-Biedl Syndrome Proteins Assemble a Coat that Traffics Membrane Proteins to Cilia. Cell. 2010, 141: 1208-1219. 10.1016/j.cell.2010.05.015.
Lechtreck K-F, Johnson EC, Sakai T, Cochran D, Ballif BA, Rush J, Pazour GJ, Ikebe M, Witman GB: The Chlamydomonas reinhardtii BBSome is an IFT cargo required for export of specific signaling proteins from flagella. J Cell Biol. 2009, 187: 1117-1132. 10.1083/jcb.200909183.
Huang K, Diener DR, Rosenbaum JL: The ubiquitin conjugation system is involved in the disassembly of cilia and flagella. J Cell Biol. 2009, 186: 601-613. 10.1083/jcb.200903066.
Sang L, Miller Julie J, Corbit Kevin C, Giles Rachel H, Brauer Matthew J, Otto Edgar A, Baye Lisa M, Wen X, Scales Suzie J, Kwong M, Huntzicker EG, Sfakianos MK, Sandoval W, Bazan JF, Kulkarni P, Garcia-Gonzalo FR, Seol AD, O'Toole JF, Held S, Reutter HM, Lane WS, Rafiq MA, Noor A, Ansar M, Devi AR, Sheffield VC, Slusarski DC, Vincent JB, Doherty DA, Hildebrandt F: Mapping the NPHP-JBTS-MKS Protein Network Reveals Ciliopathy Disease Genes and Pathways. Cell. 2011, 145: 513-528. 10.1016/j.cell.2011.04.019.
Jiang S-T, Chiou Y-Y, Wang E, Chien Y-L, Ho H-H, Tsai F-J, Lin C-Y, Tsai S-P, Li H: Essential role of nephrocystin in photoreceptor intraflagellar transport in mouse. Hum Mol Genet. 2009, 18: 1566-1577. 10.1093/hmg/ddp068.
Shiba D, Manning DK, Koga H, Beier DR, Yokoyama T: Inv acts as a molecular anchor for Nphp3 and Nek8 in the proximal segment of primary cilia. Cytoskeleton. 2010, 67: 112-119.
Li A, Saito M, Chuang J-Z, Tseng Y-Y, Dedesma C, Tomizawa K, Kaitsuka T, Sung C-H: Ciliary transition zone activation of phosphorylated Tctex-1 controls ciliary resorption, S-phase entry and fate of neural progenitors. Nat Cell Biol. 2011, 13: 402-411. 10.1038/ncb2218.
Chuang J-Z, Yeh T-Y, Bollati F, Conde C, Canavosio F, Caceres A, Sung C-H: The Dynein Light Chain Tctex-1 Has a Dynein-Independent Role in Actin Remodeling during Neurite Outgrowth. Dev Cell. 2005, 9: 75-86. 10.1016/j.devcel.2005.04.003.
Huang L, Szymanska K, Jensen VL, Janecke AR, Innes AM, Davis EE, Frosk P, Li C, Willer JR, Chodirker BN, Greenberg CR, McLeod DR, Bernier FP, Chudley AE, Müller T, Shboul M, Logan CV, Loucks CM, Beaulieu CL, Bowie RV, Bell SM, Adkins J, Zuniga FI, Ross KD, Wang J, Ban MR, Beck C, Nürnberg P, Douglas S, Craft CM, Akimenko M-A, et al: TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am J Hum Genet. 2011, 89: 713-730. 10.1016/j.ajhg.2011.11.005.
Khanna H, Davis EE, Murga-Zamalloa CA, Estrada-Cuzcano A, Lopez I, den Hollander AI, Zonneveld MN, Othman MI, Waseem N, Chakarova CF, Maubaret C, Diaz-Font A, MacDonald I, Muzny DM, Wheeler DA, Morgan M, Lewis LR, Logan CV, Tan PL, Beer MA, Inglehearn CF, Lewis RA, Jacobson SG, Bergmann C, Beales PL, Attié-Bitach T, Johnson CA, Otto EA, Bhattacharya SS, Hildebrandt F, et al: A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat Genet. 2009, 41: 739-745. 10.1038/ng.366.
Acknowledgments
The authors would like to acknowledge funding from the European Community's Seventh Framework Programme FP7/2009 under grant agreement no: 241955, SYSCILIA. http://syscilia.org/
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare no competing financial interests.
Authors' contributions
KS and CAJ contributed equally to the preparation of the manuscript. Both authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Szymanska, K., Johnson, C.A. The transition zone: an essential functional compartment of cilia. Cilia 1, 10 (2012). https://doi.org/10.1186/2046-2530-1-10
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2046-2530-1-10