
Abstract
Trypsin was purified from the digestive system of carp Catla catla (Hamilton) by ammonium sulfate fractionation, diethylaminoethyl-cellulose column chromatography, and Benzamidine Sepharose 4 fast flow column affinity chromatography. Trypsin was purified 26.2-fold with an 11.1% yield. The purified enzyme was active between pH 7.0 and 9.8, and maximal activity of the enzyme was observed at pH 7.0. Highest activity was found at 40°C. The activity was reduced to 52.84% at 60°C and was completely lost at 70°C. An addition of 2 mM CaCl2 enhanced trypsin activity during the 8-h incubation. The Km, Kcat, and catalytic efficiency values of purified enzyme were 0.062 mM and 19.23/s, and 310.16/s/mM, respectively. The enzyme activity was inhibited by soybean trypsin inhibitor, phenylmethylsulfonylflouride, and N-α-p-tosyl-l-lysine chloromethyl ketone. The molecular mass of the purified enzyme was 20.2 kDa by sodium dodecyl sulfate polyacrylamide gel electrophoresis. Mass spectrometry study of purified enzyme gave the peptide sequences LGEHNIAVNEGTEQFIDSVK (MW = 2,027.9568) and HPSYNSRNLDNDIM (MW = 1,692.6952) showing identical sequence with trypsin from various fishes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Background
Fish viscera are a potential source of digestive enzymes, especially digestive proteases. Proteases represent an important class of industrial enzymes, accounting about 50% of the total sale of the enzymes in the world (Souza et al. 2007). Various digestive proteases such as aspartic protease pepsin and serine proteases - trypsin, chymotrypsin, and elastase - are isolated from the fish viscera. Trypsin (EC 3.4.21.4) plays a pivotal role in digestive physiology. This endopeptidase hydrolyzes peptide bonds at the carboxyl end of lysine and arginine residues. Trypsin plays major roles in biological processes including digestion and activation of zymogens (Cao et al. 2000). Proteases have diverse industrial applications such as in detergent, food, pharmaceutical, leather and silk industries (Klomklao et al. 2005). Fish viscera have wide biotechnological potential as a source of digestive enzymes, especially digestive proteases that have high activity over a wide range of temperature and pH and exhibit high catalytic activity at a relatively low concentration (Shahidi and Kamil 2001;Haard 1998).
In most teleosts, trypsin is synthesized in the cells of pyloric caecum as an inactive precursor trypsinogen, which is secreted into the intestinal lumen and activated by enteroproteases (Marcuschi et al. 2010). Though trypsin and trypsin-like serine proteases have been isolated from several marine species, there is scarcity of information on the digestive proteases of tropical freshwater fishes, especially of carps. Pancreatic proteases of common carp Cyprinus carpio were purified, and their kinetic properties were studied (Cohen and Gertler 1981;Cohen et al. 1981). Two isoforms of trypsin GT-A and GT-B were isolated and purified from grass carp Ctenopharyngodon idellus (Liu et al. 2007). Trypsin was purified from the intestine of hybrid tilapia Oreochromis niloticus × O. aureus (Wang et al. 2010) and from the pyloric caecum of Amazonian tambaqui Colossoma macropomum (Marcuschi et al. 2010).
Indian major carp catla, Catla catla (Hamilton), belongs to the family Cyprinidae. This is an economically important freshwater fish species. Catla is a zooplanktivore, surface feeder. The important characteristic features of the members of the Cyprinidae are the following: the stomach and pyloric caeca are absent; the esophagus merges directly into the intestine; and there is the presence of diffused hepatopancreas. (Rathore et al. 2005) studied the digestive enzyme profile of catla during ontogenesis. They reported the appearance of less numbers of protease activity bands at the early larval stage, which gradually increased during ontogenic development. High molecular mass bands (41.8 to 127.8 kDa) appeared first, followed by low molecular mass bands (17.8 to 37.2 kDa).
No information regarding the characteristics of trypsin from catla has been reported despite the importance of this species for the Indian market. For a better understanding of the physiology and successful farming of this species and utilization of its processing wastes, more precise information about its digestive proteases is needed. Detailed information of the protease may be useful for developing new feed protein sources and improving nutritional health of the fish. The processing wastes, viscera of freshwater fishes in India, are usually discarded directly into the environment causing environmental pollution. The present investigation aimed to extract and purify trypsin from the digestive system of adult catla C. catla, and to generate basic information related to biochemical and kinetic characteristics of purified trypsin.
Methods
Maintenance of fish and collection of tissue
Indian major carp, C. catla, catla (329.17 ± 14.86 g) were collected from a local fish market in Delhi and were acclimatized at outdoor conditions for 15 days. Fish were fed with artificial diet (40% protein) consisting of dried fish powder, wheat flour, cod liver oil, and vitamin and mineral premixes (Chakrabarti et al. 2006); pH and water temperature of water ranged from 7.85 to 8.27 and from 28.5°C to 29.0°C, respectively, during experiment. Dissolved oxygen level of water was maintained above 5 mg/l with the help of aerators. Fish were kept in fasting condition for 48 h prior to sampling for complete evacuation of the digestive tract. Six fish were anesthetized with MS 222 (tricaine methanesulfonate) before sacrifice. Individual fish was dissected; the digestive system and associated glands (hepatopancreas) were removed from the body, cleaned, weighted, and immediately frozen at −20°C till further use.
Preparation of crude extract
Digestive tracts and hepatopancreas collected from six fishes were pooled; total weight of tissue was recorded and homogenized in sample buffer (10 mM Tris–HCl and 10 mM CaCl2; pH 8.0) in the ratio of 1:3 (tissue to sample buffer). Homogenized sample was passed through pretreated cheese cloth (kept in 1% ethylenediaminetetraacetate (EDTA) for 12 h to separate excess fats. The suspension was then centrifuged at 18,000 × g at 4°C for 30 min. The floating fat phase was removed, and the solution was filtered through a Buchner funnel (Borosilicate grade-2) under vacuum (ROCKER-300, TARSONS, Mumbai, India); volume of the solution was measured, and this sample was called as crude enzyme extract.
Purification of crude sample
Crude extract was subjected to ammonium sulfate fractionation (Englard and Seifter 1990). Saturated ammonium sulfate (30%) was slowly added to the crude extract with constant stirring. The sample was then centrifuged at 18,000 × g at 4°C for 30 min, and the supernatant was brought to 50% saturation by further addition of ammonium sulfate. The sample was then centrifuged at 18,000 × g at 4°C for 30 min, and the precipitate was collected, resuspended in the minimal volume of the sample buffer. The sample was dialyzed with dialysis bags (D 0530, 12.4 kDa; Sigma-Aldrich Corporation, St. Louis, MO, USA) overnight against the same buffer and was filtered through a 0.45-μm polyethersulfone membrane (25 mm in diameter; Whatman, Maidstone, England) syringe filter. Trypsin activity and protein concentration of filtrate were assayed. Filtrate was applied slowly (0.25 ml/min) to a diethylaminoethyl (DEAE)-cellulose column (0.5 × 5.5-cm column, Bio-Rad, Hercules, CA, USA). Ionic capacity of the DEAE-cellulose column is 175 μEq/ml, and dynamic binding capacity of the matrix is >30 mg bovine serum albumin (BSA)/ml. The column was washed with the equilibration buffer until the effluent had no detectable absorption at 280 nm. The whole process was carried out at 4°C.
Trypsin-like enzymes were then eluted from the column using a step gradient of NaCl with different concentrations ranging from 100 to 500 mM NaCl in the starting buffer. The flow rate was adjusted to 24 ml/h with the help of a peristaltic pump (Amersham Biosciences, Uppsala, Sweden), and 4.5-ml fractions were collected. The eluents were monitored at 280 nm for protein. For trypsin activity, the change in absorbance was measured at 410 nm using N-α-benzoyl-dl-arginine-p-nitroanalide (BAPNA) as substrate. Highest activity fractions were pooled as PF1 and were further processed with affinity chromatography. The pooled fraction was adjusted to 0.2 M KCl by the addition of solid salt and applied to a Benzamidine Sepharose 4 fast flow column (1.6 × 2.5 cm, 5 ml; Amersham Biosciences, Uppsala, Sweden). Sample was applied at 0.25 ml/min, and the column was washed at a flow rate of 20 ml/h. Fraction of 2.5 ml were collected. The unadsorbed protein was devoid of any trypsin activity. Trypsin was eluted from the column by 0.1 M acetic acid.
The contents of different tubes which showed significant trypsin activity were pooled as PF2; the pooled sample was filtered through a Whatman polyethersulfone membrane (0.45 μm). The sample was then divided into aliquots and was used for various assays.
Protein determination
Total protein content of the sample was measured by following the method of (Bradford 1976) using BSA as a standard. The absorbance was measured at 595 nm using UV-visible spectrophotometer (UV-1601, Shimadzu, Kyoto, Japan).
Specific trypsin activity
Trypsin activity was measured with BAPNA (Sigma-Aldrich Corporation, St. Louis, MO, USA). An amount of 750 μl of BAPNA (1 mM in 50 mM Tris–HCl, pH 8.2, 20 mM CaCl2) was incubated with 10 μl of enzyme extract at 37°C, and change of absorbance was recorded under kinetic mode for 3 min at 410 nm (Erlanger et al. 1961). Trypsin activity was expressed as change in absorbance per minute per milligram protein. One enzyme unit was defined as the amount of enzyme which hydrolyzed 1 mM of BAPNA per minute. Specific activity was expressed as enzyme units per milligram of protein. The molar extinction coefficient of p-nitroanalide liberated from chromogens of BAPNA is 8,800.
Optimum pH and temperature
The effect of pH on the purified enzyme was evaluated using BAPNA (1 mM) as substrate at 25°C (Castillo-Yáñez et al. 2005); pH ranged from 4 to 10.1 (0.05 M acetate-phosphate-Tris-borate buffer). The activity was measured under kinetic mode (Erlanger et al. 1961). The substrate at an appropriate pH was used as blank, and distilled water was added instead of enzyme.
For the optimum temperature study, the enzyme sample was incubated at various temperatures ranging from 10°C to 70°C without substrate for 1 h (Castillo-Yáñez et al. 2005). Trypsin activity was then measured using 1 mM BAPNA in 50 mM Tris–HCl buffer (pH 8.2, 20 mM CaCl2) as substrate under kinetic mode (Erlanger et al. 1961).
Effect of CaCl2 on thermal stability
The effect of CaCl2 on thermal stability was determined by heating the enzyme at 40°C (Klomklao et al. 2004). The enzyme sample was incubated with 50 mM Tris–HCl (pH 8.0 in the presence of 2 mM CaCl2) for time intervals of 0, 1, 2, 3, 4, 5, 6, 7, and 8 h. The residual activity of each sample was measured with BAPNA (1 mM, pH 8.2) as substrate at 25°C.
Kinetic characteristics
The Michaelis-Menten constant (Km), maximum velocity (Vmax), and catalysis constant (Kcat) were evaluated (Lineweaver and Burk 1934). The activity was assayed with varying concentrations of BAPNA ranging from 0.0156 to 2 mM. The final enzyme concentration for the assay was estimated as millimolar; Km and Vmax were evaluated by plotting the data on a Lineweaver-Burk double reciprocal graph (Prism 5 Computer Programme, Graph Pad Software, San Diego, CA, USA). Turnover number (Kcat) was calculated from the equation Kcat = Vmax/[E], where [E] is the active enzyme concentration (millimolar) and Vmax is the maximal velocity. Catalytic or the physiological efficiency of the substrate was calculated by the equation: K cat /K m .
Effect of inhibitors
Inhibition was measured according to (Garcia-Carreno et al. 1993). Purified trypsin (10 μl) was incubated with different specific protease inhibitors (10 μl), such as the serine-protease inhibitors phenylmethylsulfonylflouride (PMSF) (100 mM) and soybean trypsin inhibitor (SBTI) (250 mM), the trypsin-specific inhibitor N-α-p-tosyl-l-lysine chloromethyl ketone (TLCK) (10 mM), and the metalloprotease deactivator EDTA (20 mM). The residual activity was measured at 410 nm. Percentage activity in inhibition assays was reported, considering activity in the absence of an inhibitor as 100%.
Electrophoresis
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) of 12% polyacrylamide was carried out to evaluate the molecular mass and the purity of the enzyme sample (Laemmli 1970). Enzyme extract (20 μg protein sample) was loaded on to each well, and electrophoresis was performed (30 mA) on a vertical dual mini gel electrophoresis device (Hoefer SE-260, Amersham Pharmacia, Uppsala, Sweden) at a controlled temperature of 4°C. The gel was then washed and stained with 0.1% Coomassie brilliant blue (CBB) (Mumbai, India) in methanol/acetic acid/water (40:10:40) for 2 h. Destaining was done with the same solution without CBB for 1 h. The gel was documented in calibrated densitometer (GS-800, Bio-Rad, CA, USA) with the help of Quantity One 4.5.1 Software.
Matrix-assisted laser desorption ionization-time of flight mass spectrometry
The lyophilized enzyme sample was reconstituted (0.1 to 1 mg) in 100 μl buffer of 10 mM Tris–HCl and 10 mM CaCl2 (pH 8.0) for sequencing study. Protein (trypsin) was incubated at 37°C for 12 h in 50 mM ammoniumbicarbonate buffer. After the self cleavage of the protein into peptide, this solution was desalted using Zip-Tip C18 (Millipore Co., Billerica, MA, USA). Desalted sample was spotted with α-cyano-4-hydroxy-cinnamic acid for peptide mass finger printing and mass spectrometry (MS/MS) of peptides. Matrix-assisted laser desorption ionization-time of flight (MALDI TOF-TOF) (4800 Plus MALDI TOF/TOFTM, Applied Biosystems, LABINDIA, New Delhi, India) MS/MS data were submitted for the database search using Protein Pilot 2.0 Software (AB SCIEX, Framingham, MA, USA).
This software accepts only MS/MS spectra for the identification of protein, and protein was identified on that basis. Peptide sequence of target protein was searched for alignment using protein pilot software (Uniprot 2012).
Results and discussion
Purification of trypsin
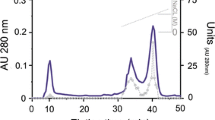
In the first step of purification, crude enzyme extract (0.7 U/mg protein) was concentrated by ammonium sulfate precipitation. The specific enzyme activity of the sample after ammonium sulfate precipitation (30% to 50%) and followed by dialysis was 3.9 U/mg protein (Table 1). Ammonium sulfate precipitation served as an initial step to remove other proteins in the crude enzyme extract. The dialysate showed 5.8-fold purification compared with the crude enzyme extract with a recovery of 41.7%. The elution profiles of the trypsin activity and proteins after filtration on a DEAE-cellulose column were shown in Figure1. Step gradient of NaCl in the range of 100 to 500 mM NaCl was applied to the DEAE-cellulose matrix. Fractions from 1 to 43 correspond to the unbound when washed with the equilibration buffer. In a 100-mM NaCl elution, more specific trypsin activity with BAPNA was found between the fractions 53 and 66. The specific activity of the pooled fraction PF1 (50 to 69) of 100-mM NaCl elution was 11.0 U/mg protein; 16.4-fold purification was found compared with the crude extract. Pooled fraction PF1 was further subjected to affinity chromatography. After the final purification in affinity chromatography, enzyme sample showed a single peak corresponding to the fraction number 44 (Figure2); 26.2-fold purified trypsin (PF2) was obtained with 11.1% recovery. The specific activity of the purified sample was 17.6 U/mg protein.

Chromatography pattern of C. catla trypsin using DEAE-cellulose column. Column was eluted with 10 mM Tris–HCl and 10 mM CaCl2 (pH 8.0) containing 100, 150, 200, and 500 mM NaCl. Main activity fractions were eluted by 10 mM Tris–HCl and 10 mM CaCl2 (pH 8.0) containing 100 mM NaCl. Elution flow rate was 24 ml/h. Content of protein is expressed as absorbance at 280 nm. Enzyme activity is expressed as absorbance at 410 nm.

Purification profile of trypsin of C. catla on Benzamidine Sepharose 4 fast flow column. Trypsin was eluted using 0.1 M acetic acid. The fractions eluted from the column were immediately adjusted to pH 8.0 by adding 700 μl of 1 M Tris–HCl buffer (pH 9.0). Elution flow rate was 20 ml/h. Content of protein is expressed as absorbance at 280 nm. Enzyme activity is expressed as absorbance at 410 nm.
In grass carp, 2.2-fold purification of enzyme with 43.6% yield was found after ammonium sulfate precipitation (Liu et al. 2007). Two isoforms of trypsin, GT-A and GT-B, were purified. The specific activity of GT-A increased from 0.4 to 86.7, and the purification fold increased from 1.0 to 201.7, whereas the specific activity of GT-B increased from 0.4 to 60.1, and the purification fold increased from 1.0 to 139.7. (Cohen et al. 1981) obtained 17% yield after purification of the common carp trypsin. A 60-fold purification of alkaline protease was obtained from the intestine of Clarias batrachus by ion exchange chromatography on DEAE-cellulose column (Mukhopadhyay et al. 1978).
Optimum pH and temperature
The effect of pH on enzyme activity was determined over a pH range of 4.0 to 10.1. The purified enzyme was active between pH 7.0 and 9.8. Most enzymes suffer irreversible denaturation in very acidic and alkaline conditions, with low activity found at very acidic and high alkaline pH. A similar result was found with catla. The lowest activity was found at pH 4.0, while the highest activity was observed at pH 7.0 (Figure3). A 47.36% loss of enzyme activity was found at pH 10.1. The optimum pH of two isoforms of trypsin obtained from grass carp were 8.0 (GT-A) and 8.5 (GT-B); optimum pH for hybrid tilapia trypsin was 9.0 (Liu et al. 2007; Wang et al. 2010). Two particular features were observed in carp enzymes, instability in low pH and trypsin showing as an anionic protein (Cohen and Gertler 1981;Cohen et al. 1981). Protease purified from the intestine of C. batrachus showed optimum activity at pH 8.0 (Mukhopadhyay et al. 1978).

Effect of pH on enzyme activity. Activity was measured at various pH values ranging from 4.0 to 10.1 using 1 mM BAPNA as substrate at 25°C. Percentage of enzyme activity was estimated considering 100% - the highest activity detected in the assay.
The purified trypsin was incubated at various temperatures to study the effect of temperature on enzyme activity (Figure4). The enzyme activity at 10°C was 76.75% (5.78 U/mg protein) as compared to the activity found at 40°C. The activity increased with the increasing temperature. The highest activity was found at 40°C (7.54 U/mg protein). Activity gradually decreased with the increase of temperature. The activity was reduced to 52.84% (3.9834 U/mg protein) at 60°C and was almost completely lost at 70°C (0.0053 U/mg protein). A direct correlation was found between the temperature of the fish habitat and the thermal stability of trypsin (Kishimura et al. 2008). Trypsins from tropical fish showed higher thermal stability compared with those in fish that adapted to cold environment. This may be due to lesser hydrophilicity and stronger hydrophobic interactions in the protein center (Klomklao et al. 2007 2009). Purified trypsin of catla showed increased activity up to 40°C, and then the activity gradually reduced. The activity was significantly reduced at a temperature of 70°C. This may be due to thermal denaturation. In grass carp, the optimum temperature for two isoforms of trypsin GT-A and GT-B were 38.5°C and 44°C, respectively (Liu et al. 2007). Catla trypsin may have a potential application value where low processing temperature and higher enzyme activity is required.

Optimum temperature for trypsin activity. Activity was assayed at various temperatures ranging from 10°C to 70°C using 1 mM BAPNA as substrate (pH 8.2). Percentage of enzyme activity was estimated based on the highest activity detected in the assay as 100%.
Effect of CaCl2 on thermal stability
Incubation of purified trypsin with 2 mM CaCl2 showed increased activity up to 8 h (Figure5). The enzyme activity was 11.7% higher after 4 and 8 h of incubation compared to the initial value. In Greenland cod Gadus ogac, the gastric proteinase activity was increased in the presence of CaCl2 (Squires et al. 1986). Trypsin of common carp Cyprinus carpio was stable at pH 5 to 9 in the presence of 0.1 M CaCl2 at 4°C for at least 1 week (Cohen and Gertler 1981). (Liu et al. 2007) found that the activity of two isoforms of trypsin of grass carp was not affected substantially by Ca+2.

Effect of calcium ion on the stability of trypsin of C. catla. The enzyme was incubated with 2 mM of CaCl2 at 40°C and a pH of 8.0 for 0 to 8 h. The activity was measured using 1 mM BAPNA as substrate at 25°C and pH 8.2. Percentage of enzyme activity was estimated, considering 0 h activity as 100%.
Kinetic characteristics
The Km value of trypsin was 0.062 mM (Figure6). Kcat value was calculated as 19.23/s. The catalytic efficiency of the purified trypsin was 310.16/s/mM. The Km and Kcat values and the catalytic efficiency of catla in the present study were higher compared with that of the common carp. In the common carp, the Km and Kcat values and the catalytic efficiency were 0.039 mM, 3.10/s, and 79.5/s/mM, respectively (Cohen et al. 1981).

Lineweaver-Burk plot for trypsin kinetics. 1/[V] and 1/[S] represent the reciprocal of velocity and substrate, respectively.
Effect of inhibitors
Proteases can be classified by their sensibility to various inhibitors (Ktari et al. 2012). Trypsin activity was completely inhibited by the serine protease inhibitors, SBTI and PMSF, and the specific inhibitor of trypsin, TLCK. These results suggested that the purified enzyme is a serine protease and classified as trypsin-like enzyme. The metalloprotease inhibitor EDTA inhibited 59.53% of the enzyme activity. This shows the importance of ions in enzyme activity. A similar result was found in zebra blenny Salaria basilisca (Ktari et al. 2012) and in grass carp (Liu et al. 2007). (Jany 1976) reported the presence of seryl-protease trypsin in stomachless Carassius auratus gibelio (Bloch).
Purity and molecular mass
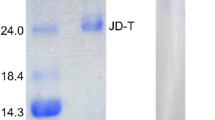
The purified enzyme (PF2) showed a single band on SDS-PAGE (Figure7). The molecular mass of the band was 20.2 kDa. The presence of a single band in SDS-PAGE confirmed the purity of the sample. In the common carp, the molecular mass of trypsin was approximately 25.0 kDa (Cohen and Gertler 1981). The relative molecular masses of two isoforms of trypsin GT-A and GT-B were 30.74 and 26.4 kDa, respectively (Liu et al. 2007). The molecular mass of trypsin of hybrid tilapia was 22.0 kDa (Wang et al. 2010). In freshwater fish, tambaqui (Colossoma macropomum), the purified enzyme showed a single band of 27.5 kDa (Marcuschi et al. 2010).

SDS-PAGE of enzyme sample of C. catla at various stages of purification. The sample was diluted (1:1) with the sample buffer. M, molecular marker comprises phosphorylase b (97,400), bovine albumin (66,200), ovalbumin (45,000), carbonic anhydrase (31,000), trypsin inhibitor (21,500), and lysozyme (14,400). After electrophoresis, the gel was stained with CBB for 2 h and was destained. CE, crude extract; IEC, purified fraction obtained by ion exchange chromatography; AF, purified fraction obtained by affinity chromatography.
MALDI-TOF mass spectrometry
The peptide sequence of the purified trypsin was obtained by MALDI-TOF/TOF mass spectrometry. Two peptide sequences with a 99% confidence score were obtained. The peptide sequences obtained from catla were LGEHNIAVNEGTEQFIDSVK (MW = 2,027.9568, MS/MS mass error = 0.0020) and HPSYNSRNLDNDIM (MW = 1692.6952, MS/MS mass error = −0.0097). All these peptides were mapped to the anionic trypsin of fish. Peptides obtained in the MS/MS ion search have arginine or lysine residue, which is a characteristic feature of trypsin-digested peptides. The protein was identified as trypsin (EC 3.4.21.4) showing similarity with trypsin obtained from Salmo salar (percentage coverage = 14.3%). The peptide sequence of catla was aligned and compared with the sequences of other fishes (Table 2) using UniProtKB BLAST (Uniprot 2012).
Conclusions
The present investigation has established some important biochemical properties of trypsin purified from the digestive system of catla. The protease had a similarity with trypsin from other fishes. Stability at high pH and low temperature indicates the potential application of this protease in detergent and in the food industry. Enzymes extracted from the fish viscera (the waste part of the fish body) may be used in the food processing industry and thus making beneficial and productive use of the fish processing wastes.
References
Bradford MM: A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976, 72: 248–254. 10.1016/0003-2697(76)90527-3
Cao MJ, Osatomi K, Sujuki M, Hara K, Tachibana K, Ishihara T: Purification and characterization of two anionic trypsins from the hepatopancreas of carp. Fish Sci 2000, 66: 1172–1179. 10.1046/j.1444-2906.2000.00185.x
Castillo-Yáñez FJ, Pacheco-Aguilar R, García-Carreño FL, Navarrete-Del Toro MA: Isolation and characterization of trypsin from pyloric caeca of Monterey sardine Sardinops sagax caerulea. Com Biochem Physiol 2005, 140B: 91–98.
Chakrabarti R, Rathore MS, Kumar S, Mittal P: Functional changes in digestive enzymes and characterization of proteases of silver carp (male) and bighead carp (female) hybrid, during ontogeny. Aquaculture 2006, 253: 694–702. 10.1016/j.aquaculture.2005.08.018
Cohen T, Gertler A: Pancreatic proteolytic enzymes from carp (Cyprinus carpio) I. Purification and physical properties of trypsin, chymosin, elastase and carboxypeptidase B. Comp Biochem Physiol B 1981, 98: 517–521.
Cohen T, Gertler A, Birk Y: Pancreatic proteolytic enzymes from carp (Cyprinus carpio) I. Purification and physical properties of trypsin, chymotrypsin, elastase and carboxipeptidase B. Comp Biochem Physiol 1981, 69B: 639–646.
Englard S, Seifter S Duetscher MP (ed) Methods in enzymology. In Precipitation techniques. Guides to protein purification, vol. 182. Academic Press Inc, San Diego; 1990:285–300.
Erlanger BF, Kokowsky N, Cohen W: The preparation and properties of two new chromogenic substrates of trypsin. Arch Biochem Biophy 1961, 95: 271–278. 10.1016/0003-9861(61)90145-X
Garcia-Carreno FL, Dimes LE, Haard NF: Substrate-gel electrophoresis for composition and molecular weight of proteinases or proteinaceous proteinases inhibitors. Anal Biochem 1993, 214: 65–69. 10.1006/abio.1993.1457
Haard NF: Specialty enzymes from marine organisms. Food Technol 1998, 52: 64–67.
Jany KD: Studies on the digestive enzymes of the stomachless bonefish Carassius auratus gibelio (Bloch): endopeptidases. Comp Biochem Physiol 1976, 53B: 31–38.
Kishimura H, Klomklao S, Benjakul S, Chun BS: Characteristics of trypsin from the pyloric caeca of walleye Pollock (Theragra chalcogramma) . Food Chem 2008, 106: 194–199. 10.1016/j.foodchem.2007.05.056
Klomklao S, Benjakul S, Visessanguan W: Comparative studies on proteolytic activity of spleenic extract from three tuna species commonly used in Thailand. J Food Biochem 2004, 28: 355–372. 10.1111/j.1745-4514.2004.05203.x
Klomklao S, Benjakul S, Visessanguan W, Simpson BK, Kishimura H: Partitioning and recovery of proteinase from tuna spleen by aqueous two-phase systems. Process Biochem 2005, 40: 3061–3067. 10.1016/j.procbio.2005.03.009
Klomklao S, Benjakul S, Visessanguan W, Kishimura H, Simpson BK: Purification and characterisation of trypsins from the spleen of skipjack tuna (Katsuwonus pelamis) . Food Chem 2007, 100: 1580–1589. 10.1016/j.foodchem.2006.01.001
Klomklao S, Kishimura H, Nonami Y, Benjakul S: Biochemical properties of two isoforms of trypsin purified from the intestine of skipjack tuna (Katsuwonus pelamis) . Food Chem 2009, 115: 155–162. 10.1016/j.foodchem.2008.11.087
Ktari N, Khaled HB, Nasri R, Jellouli K, Ghorbel S, Nasri M: Trypsin from zebra blenny (Salaria basilisca) viscera: purification, characterisation and potential application as a detergent additive. Food Chem 2012, 130: 467–474. 10.1016/j.foodchem.2011.07.015
Laemmli UK: Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227: 680–685. 10.1038/227680a0
Lineweaver H, Burk D: The determination of enzyme dissociation constants. J Am Chem Soc 1934, 56: 658–666. 10.1021/ja01318a036
Liu ZY, Wang Z, Xu SY, Xu LN: Two trypsin isoforms from the intestine of grass carp (Ctenopharyngodon idellus) . J Comp Physiol B 2007, 177: 655–666. 10.1007/s00360-007-0163-6
Marcuschi M, Esposito TS, Machado MFM, Hirata IY, Machado MFM, Silva MV, Carvalho LB, Oliveira V, Bezerra RS: Purification, characterization and substrate specificity of a trypsin from the Amazonian fish tambaqui (Colossoma macropomum) . Biochem Biophys Res Commun 2010, 396: 667–673. 10.1016/j.bbrc.2010.04.155
Mukhopadhyay PK, Dehadrai PV, Banerjee SK: Studies on intestinal protease: purification and effect of dietary proteins on alkaline protease activity of the air-breathing fish, Clarias batrachus (Linn.). Hydrobiologia 1978,57(I):11–15.
Rathore RM, Kumar S, Chakrabarti R: Digestive enzyme patterns and evaluation of protease classes in Catla catla (Family: Cyprinidae) during early developmental stages. Comp Biochem Physiol B 2005, 142: 98–106. 10.1016/j.cbpc.2005.06.007
Shahidi F, Kamil YVAJ: Enzymes from fish and aquatic invertebrates and their application in the food industry. Trends Food Sci Tech 2001, 12: 435–464. 10.1016/S0924-2244(02)00021-3
Souza AAG, Amaral IPG, Santo ARE, Carvalho LB Jr, Bezerra RS: Trypsin-like enzyme from intestine and pyloric caeca of spotted goatfish (Pseudupeneus maculatus) . Food Chem 2007, 100: 1429–1434. 10.1016/j.foodchem.2005.12.016
Squires EJ, Haard NF, Felthem LA: Gastric proteases of the Greenland cod Gadus ogac . II. Structural properties. Biochem Cell Biol 1986, 64: 215–222.
Uniprot [Universal Protein Resource]: Sequence or Uniprot identifier. 2012. http://www.uniprot.org/blast/
Wang Q, Gao ZX, Zhang N, Shi Y, Xie XL, Chen QX: Purification and characterization of trypsin from the intestine of hybrid tilapia (Oreochromis niloticus × O. aureus) . J Agri Food Chem 2010, 58: 655–659. 10.1021/jf903052s
Acknowledgements
The authors are thankful to the Department of Science and Technology, New Delhi for the financial support. Mass spectrometry study was conducted at the Central Instrumentation Facility, South Campus, University of Delhi.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RC has conceived the project, designed the experiment, analyzed data, and has maximum contribution in writing the manuscript. KSYVR has standardized some techniques as well as conducted experiments. BKK searched literature, has drawn figures, and conducted experiments. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Khangembam, B.K., YVR, K.S. & Chakrabarti, R. Purification and characterization of trypsin from the digestive system of carp Catla catla (Hamilton). Int Aquat Res 4, 9 (2012). https://doi.org/10.1186/2008-6970-4-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2008-6970-4-9