
Abstract
Duane retraction syndrome (DRS) is a neuromuscular dysfunction of the eyes. Although many causative genes of DRS have been identified in Europe and the United States, few reports have been published in regard to Chinese DRS. The aim of the present study was to explore the genetic defect of DRS in a Chinese family. Exome sequencing was used to identify the disease-causing gene for the two affected family members. Ophthalmic and physical examinations, as well as genetic screenings for variants in chimerin 1 (CHN1), were performed for all family members. Functional analyses of a CHN1 variant in 293T cells included a Rac-GTP activation assay, α2-chimaerin translocation assay, and co-immunoprecipitation assay. Genetic analysis revealed a NM_001822.7: c.637T > G variant in the CHN1 gene, which resulted in the substitution of a highly conserved C1 domain with valine at codon 213 (NP_001813.1: p.(Phe213Val)) (ClinVar Accession Number: SCV001335305). In-silico analysis revealed that the p.(Phe213Val) substitution affected the protein stability and connections among the amino acids of CHN1 in terms of its tertiary protein structure. Functional studies indicated that the p.(Phe213Val) substitution reduced Rac-GTP activity and enhanced membrane translocation in response to phorbol-myristoyl acetate (PMA). Together with previous studies, our present findings demonstrate that CHN1 may be an important causative gene for different ethnicities with DRS.
Similar content being viewed by others


Introduction
Duane retraction syndrome (DRS) is a congenital disorder that impairs eye movement1 and is caused by a failure of the sixth cranial nerve (i.e., the abducens nerve). DRS is characterized by a restriction or absence of abduction and/or adduction, partial closure of the eyelids, and globe retraction into the orbit upon adduction1. DRS is estimated to constitute 1–5% of all strabismus and to affect 0.1% of the general population2. Without treatment, DRS can result in amblyopia and blindness during childhood. Familial DRS is mainly inherited in an autosomal-dominant mode, which accounts for 10% of all DRS disorders3.
Based on the current understanding of the genetics of DRS, at least six genes—spalt like transcription factor 4 (SALL4), chimerin 1 (CHN1), homeobox A1 (HOXA1), MAF bZIP transcription factor B (MAFB), kinesin family member 21A (KIF21A), and tubulin beta 3 class III (TUBB3)—have been identified to be associated with DRS4,5. Duane retraction syndrome 2 (DURS2, OMIM 604356) and DURS3 (OMIM 617041) have been identified to be caused by variants in CHN1 and MAFB, while DURS1 (OMIM 126800) maps to chromosome 8q13. Variants in CHN1 hyper-activating α2-chimaerin are involved in familial non-syndromic DURS2 with incomplete penetrance6. CHN1 can encode α2-chimaerin, which is a Rac guanosine triphosphatase (GTPase)-activating protein (RacGAP) that functions as a phorbol ester receptor and ras-related p21-rac that can be activated by upstream signaling in the plasma membrane7,8,9. Upon activation, Rac-GTP is hydrolyzed to inactive Rac-GDP via the α2-chimaerin GAP domain10, which alters oculomotor axons and extraocular muscle dynamics during the development of the chick embryo6.
Human populations with different ethnicities and from distinct geographical locations confer a diverse overall genetic background. More than 10 CHN1 variants have been reported to be associated with DURS2 in Europe and the United States6,15. However, there are currently no reports regarding genetic abnormalities underlying DRS2 in a Chinese population. Hence, a further spectrum of variation for DRS is needed, particularly in Asian populations. The purpose of the present study was to screen for variants in DRS causal genes in a Han Chinese DRS family and to explore the functional mechanism of a novel CHN1 substitution, NM_001822.7: c.637T > G, NP_001813.1: p.(Phe213Val) (ClinVar Accession Number: SCV001335305), underlying DRS2.
Results
Clinical phenotypes and laboratory analysis
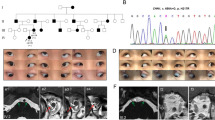
There were 37 members in the Han Chinese family who were considered for the present study, with 11 members being diagnosed with DRS during childhood. The DRS patients in this family were consistent with being representative of an incomplete dominant genetic model of DRS (Fig. 1A). We also evaluated the clinical phenotypes for 25 individuals in this family (Table 1). Among them, eight members were diagnosed with DRS and exhibited abnormal corneal optical reflections, squint angles, and globe retractions. Six family members (III:2, III:7, III:17, IV:1, IV:5, and IV:6) had bilateral DRS, whereas two individuals (II:3 and II:11) had left DRS. Interestingly, five of the DRS members were diagnosed with DRS type I, whereas one family member (III:2) was diagnosed with DRS type II. Additionally, two other family members (IV:1 and IV:5) had different DRS types in their left and right eyes.

Pedigree of DRS family with a CHN1 variant, sequencing chromatogram, and diagnostic positions of gaze analysis. (A) Pedigree of DRS family with a CHN1 variant. The proband is marked by an arrow, black symbols denote affected members, white symbols denote unaffected members, squares denote males, and circles denote females. The presence or absence of the CHN1 substitution (c.637T > G) is indicated by an ‘M’ or ‘+’, respectively. (B) Sequencing chromatogram. (C) Diagnostic positions of the gaze of subject IV:6. RefSeq of CHN1: NM_001822.7.
One family member (IV:6) was diagnosed with bilateral DRS as a proband (Fig. 1C). He exhibited bilateral horizontal-rectus muscle co-contraction and a bilateral-exodeviation limitation. The abduction limitation was mild in the right eye and moderate in the left eye. His corrected visual acuities of the right eye and left eye were each 1.0 logMAR. The results of the cornea/lens examination and fundus examination were each normal. Magnetic resonance imaging was not obtained. The degree of strabismus was + 75Δ, and stereopsis was deficient. The patient then underwent strabismus surgery. After surgery, his eye position was notably improved.
Another family member (IV:5) was diagnosed as having bilateral DRS (Supporting Information Figure S1). His bilateral eyes were mildly exodeviated. He had a history of a left head tilt since early childhood. His corrected visual acuities of the right eye and left eye were each 0.9 logMAR. The results of the cornea/lens examination and fundus examination were each normal. Magnetic resonance imaging was not obtained. Further examination revealed right hypertropia, as well as a large deorsumversion deficit. The degree of strabismus was R\L30Δ, and stereopsis was deficient. After surgery, both his left head tilt and eye position were notably improved.
Genetic analysis
DNA samples from 25 living family members from the included Han Chinese family were used for genetic studies and were comprised of samples from nine DRS patients and 16 unaffected family members. In addition, we collected 200 DNA samples from ethnically matched controls to verify CHN1 variants. Samples from two family members (III:11 and III:17) were used for exome sequencing to screen for variants. After filtering procedures (Tables S1 and S2), we identified a novel non-synonymous potential disease-associated variant (NM_001822.7: c.637T > G, NP_001813.1: p.(Phe213Val)) in CHN1 (i.e., the gene encoding α2-chimaerin). Based on a dominant model, we selected rare variants to perform co-segregation analysis for this pedigree. This variant co-segregated with all DRS family members and four individuals whose eyes were clinically normal (Fig. 1A and Table 1).
In this variant, valine was substituted for phenylalanine at position 213 in CHN1 (p.(Phe213Val)), which was not yet present in the 1000 Genomes Project dataset (https://browser.1000genomes.org/index.html). This variant was also not detected in the 200 ethnically matched controls from the same Southwestern Chinese population (data not shown). We also sequenced all coding exons and splice sites of CHN1; however, we did not detect any suspicious variations other than the c.637T > G substitution.
The p.(Phe213Val) substitution mapped to the C1 domain of CHN1, which was within its coil structure (Supporting Information Figure S2) and was located in a PKC phosphorylation site (PS00005). The p.(Phe213Val) substitution led to a highly conserved amino-acid change, as indicated by evolutionary-conservation analysis (Fig. 2). The results of MutationTaster, Sorting Tolerant From Intolerant (SIFT), and Polymorphism Phenotyping v2 (PolyPhen-2) predicted that the p.(Phe213Val) substitution had a disease-causing effect. Protein-function prediction suggested that the F213 in the wild-type CHN1 protein was associated with two other residues (R214 and Q232) via three connecting hydrogen bonds, while the substitution that we identified for CHN1 (V213) led to one connecting hydrogen bond with residue Q232. This replacement was predicted to result in the loss of two structuring H-bonds, introducing conformational changes in the affected region. For computational-stability analysis, we used the three-dimensional protein structure of human CHN1 (PBD ID: 3CXL) to assess the stability of the p.(Phe213Val) variant. The results were consistent with those of nine other variants that have been reported previously (Table 2); specifically, p.(Phe213Val) induced notable destabilization with a large decline in Gibbs free energy, as indicated by negative ΔΔG values determined by four different types of software.

CHN1 variant analysis. (A) Two-dimensional representations of the α2-chimaerin protein. The protein had three domains: C1, Rac-GAP, and Src homology 2 (SH2). The novel c.637T > G substitution altered residue F213 (p.(Phe213Val)), which was present in the C1 domain of α2-chimaerin (red) and was within a coil structure and a PKC phosphorylation site. Previously reported variants are also indicated. (B) The α2-chimaerin protein alignment among 12 different species demonstrates the evolutionary conservation of residue F213 (boxed in red). (C) Homology modeling of the CHN1 protein shows the conformational changes induced with and without the p.(Phe213Val) substitution. RefSeq of CHN1: NM_001822.7.
Functional analysis of the p.(Phe213Val) amino-acid substitution
To verify potential effects of the p.(Phe213Val) substitution on CHN1 function, we performed overexpression of variant or wild-type CHN1 to induce differential Rac-GTP activation, α2-chimaerin translocation, and self-assembly.
We used a Rac-GTP activation assay to analyze Rac-GTP levels. Compared with that of an empty vector, overexpression of wild-type CHN1 lowered Rac-GTP levels, which is consistent with the known function of α2-chimaerin. Moreover, overexpression of variant CHN1 further reduced Rac-GTP levels compared to that of wild-type CHN1 (Fig. 3A,B, and Supporting Information).

Functional studies of the novel c.637T > G CHN1 substitution (MT) compared with wild-type (WT) CHN1. (A) Western blotting analysis of Rac-GTP, V5, total Rac, and β-actin. (B) Grayscale results of Western blotting. Top blot: WT α2-chimaerin reduced Rac-GTP levels compared to those with the empty vector, and MT α2-chimaerin lowered these levels below those of WT. The levels of total Rac (second blot) and V5-α2-chimaerin (third blot) in all groups were equal, as were the levels of β-actin (bottom blot) (n = 3). (C) Western-blotting analysis of α2-chimaerin from the PMA-stimulated α2-chimaerin membrane-translocation experiment and (D) grayscale results of Western blotting. Although total α2-chimaerin was comparable in all conditions, PMA stimulated translocation of α2-chimaerin into the pelleted (membranous) fraction. MT α2-chimaerin translocation was enhanced compared to that of WT (n = 3). (E) Western blotting showing a slight increase in the self-assembly of WT α2-chimaerin with MT α2-chimaerin compared to self-assembly of WT with WT α2-chimaerin (n = 3) and (F) Grayscale results of Western blotting. Abbreviations are as follows: −: non-transfected vector, +: transfected vector WT: wild type, MT: variant type, E-W: empty vector and wild type vector, W-W: wild type vector and wild type vector, M-W: variant type vector and wild type vector. RefSeq of CHN1: NP_001813.1.
To assess the influence of the p.(Phe213Val) substitution on protein translocation from the cytoplasm to the plasma membrane, we quantified wild-type and variant α2-chimaerin in pelleted and soluble fractions prior to or after stimulation with PMA. As predicted, after PMA stimulation, variant α2-chimaerin was significantly increased in the soluble fraction, whereas it was concomitantly decreased in the pelleted fraction. For wild-type α2-chimaerin, there was no difference in the proportion of α2-chimaerin in soluble/pelleted fractions prior to and after stimulation with PMA (Fig. 3C,D, and Supporting Information). These results suggest that this α2-chimaerin variant induced aberrant translocation to the plasma membrane after PMA stimulation.
We also tested if variant hyper-activated α2-chimaerin could interact with the wild-type protein to recruit wild-type α2-chimaerin to the plasma membrane, which may further reduce Rac activity. In the presence of PMA, the immunoprecipitation of wild-type α2-chimaerin with variant α2-chimaerin was slightly enhanced compared to its interaction with itself, but this difference was not statistically significant (Fig. 3E,F, and Supporting Information).
Taken together, the p.(Phe213Val) substitution reduced Rac-GTP activity and enhanced translocation to the plasma membrane by putatively changing the conformation of α2-chimaerin in response to PMA, without affecting protein self-assembly.
American College of Medical Genetics (ACMG) evaluation of the CHN1 c.637T > G (p.(Phe213Val)) substitution
Conforming to the classification of the American College of Medical Genetics (ACMG) for evaluating the pathogenicity of different variants, the p.(Phe213Val) substitution of CHN1 was “pathogenic” as it was well-established via in-vitro functional studies supportive of a deleterious effect on the CHN1 protein (PS3) in a PKC phosphorylation site (PM1) that was absent in population databases (PM2), and there was cosegregation with affected family members (PP1), computational evidence showed a damaging effect (PP3), and a highly specific disease phenotype (DRS) with via a single gene (CHN1) (PP4). Thus, according to the ACMG evaluation, the p.(Phe213Val) substitution of CHN1 had one strong (PS3), two moderate (PM1, PM2), and more than two supportive (PP1, PP3, PP4) findings of pathogenicity, meeting the criteria of the ACMG for a substitution to be considered a pathogenic variant.
Discussion
Patients with DRS have globe retraction upon adduction, which is due to the absence of normal functioning of abducens motor neurons and the abducens nerve to regulate the lateral rectus muscle11; hence, DRS involves abnormal neuromuscular regulation of oculomotor function. DRS is divided into three types (DRS type I, DRS type II, and DRS type III) based on different muscular limits (abduction, adduction, or both, respectively) according to the Huber’s classification system12. All three types of DRS were detected in our pedigree, which is consistent with previous observations that have suggested phenotypic diversity of DRS12,13. According to genetic heterogeneity, DURS1 maps to chromosome 8q13, whereas DURS2 and DRS3 are caused by variants in the CHN1 gene and MAFB gene, respectively5,11,14.
CHN1 is one of the most important and common pathogenic genes for DRS, and nearly 10 variants (L20F, I126M, P141L, Y143H, Y148F, A223V, G228S, P252Q/S, and E313K) have already been reported (Fig. 2A)6,15,16. Previous studies on the functional analyses of these CHN1 variants have revealed that p.F213V-, L20F-, Y143H-, Y148F-, A223V-, and P252Q-α2-chimaerin mutant proteins can enhance Rac-GAP activity and membrane translocation by destabilizing the closed conformation of α2-chimaerin in response to PMA6,15,16. Additionally, in the presence of PMA, previous studies have demonstrated that the interaction of wild-type-α2-chimaerin with mutants (L20F, I126M, Y143H, Y148F, A223V, and P252Q) is significantly enhanced compared to its interaction with itself, but that there is no effect regarding the interactions of variants of α2-chimaerin (G228S and E313K)6,15,16.
These variants have primarily been detected in non-Chinese individuals6,15,16. In the present study, we reported a novel p.(Phe213Val) substitution in a large Han Chinese family with reduced penetrance to congenital DRS. We used exome sequencing and bioinformatic analysis to identify a specific p.(Phe213Val) substitution in CHN1, which was carried by all affected members in our pedigree. The p.(Phe213Val) substitution is a rare CHN1 variant that was not found in either existing databases or in the 200 ethnically matched controls analyzed in the present study. The p.(Phe213Val) substitution is a conserved amino-acid substitution (Fig. 2) and is located in a PKC phosphorylation site (PS00005), which may change its phosphorylated state. The p.(Phe213Val) substitution reduced connections between amino acids of CHN1 in terms of its tertiary protein structure (Fig. 2B). These changes were also confirmed via mutation-stability analysis.
Mutation-stability analysis was executed for the p.(Phe213Val) substitution and for the 10 other related DRS variants in CHN1 that have been reported in previous studies6,15,16. Aside from the p.P141L substitution, the p.(Phe213Val) substitution and the other nine variants were all classified as destabilizing in terms of exhibiting declines in Gibbs free energy (Table 2). Interestingly, among these variants, the p.(Phe213Val) substitution was the most unstable, as indicated by its ΔΔG value = − 2.13 via DUET software analysis. This result was consistent with the high conservation values of aa.F213, which suggests an essential contribution of aa.F213 to functional and/or structural CHN1 stability.
Based on the above analysis, we speculated that the p.(Phe213Val) substitution would induce hyper-activated α2-chimaerin RacGAP activity by destabilizing its closed conformation. In our present study, in response to PMA, α2-chimaerin with the p.(Phe213Val) substitution enhanced RacGAP activity and membrane translocation, which may have been induced by substitution-mediated opening of the closed conformation of CHN1. Interestingly, p.L20F-, Y143H-, Y148F-, A223V-, and P252Q-α2-chimaerin mutant proteins appear to enhance membrane translocation and RacGAP activity by destabilizing the closed conformation of α2-chimaerin in response to PMA, as indicated by results from previous studies6,15.
However, in our present study, in the presence of PMA, the interaction of wild-type-α2-chimaerin with mutants (L20F, I126M, Y143H, Y148F, A223V, and P252Q) was significantly enhanced compared to its interaction with itself; however, variants (G228S and E313K) had no such changes in their interactions with each other6,15. Consistent with the results of the interactions between mutants (G228S and E313K), we detected that the interaction between the p.(Phe213Val) substitution and wild-type-α2-chimaerin was not significantly enhanced. This may account for the reduced penetrance found in this pedigree, which would be consistent with the known incomplete penetrance in G228S and E313K variant pedigrees. Taken together, our findings suggest that α2-chimaerin with p.F231V can enhance membrane translocation and RacGAP activity.
In the family analyzed in the present study, the p.F231V in CHN1 excessively activated α2-chimaerin via enhancing α2-chimaerin’s membrane translocation and activity, hydrolyzing Rac-GTP, and altering oculomotor axons and extraocular muscle dynamics, all of which led to the DURS2 phenotype. To the best of our knowledge, the present study is the first to report that a substitution in the CHN1 gene is involved in the pathogenesis of DRS in a Chinese family. Combined with the results of previous studies, our present study corroborates that CHN1 plays an important role in the pathogenesis of DRS in humans.
Materials and methods
Ethical considerations
This study was approved by the Clinical Research Ethics Committee of the Second Hospital of Yunnan Province (2019098) and adhered to the Declaration of Helsinki Principles. Written informed consent for both study participation and publication of identifying information/images in an online open-access publication was obtained from all participants or their parents (for participants under the age of 18 years).
Human subjects
A four-generation Han Chinese family diagnosed with autosomal incompletely dominant congenital DRS was enrolled in the present study at the Second People’s Hospital of Yunnan Province (Kunming, China). This family included 34 subjects comprised of 10 affected members and 24 unaffected members. In the present study, we recruited 25 of these family members, which included eight affected members (Fig. 1). The 200 ethnically matched controls had normal eyes and were also enrolled at the Second People’s Hospital of Yunnan Province (Kunming, China).
All 25 family members underwent physical and ophthalmic examinations, which included tests of vision, slit-lamp biomicroscopy, and fundus examination during pupil dilation, as well as measurements of diopter, degree of strabismus, eye movement, and corneal curvature. These tests and measurements were used to identify whether there were any other ocular or systemic abnormalities present in the included family members (Table 1).
Exome sequencing and data analysis
Genomic DNA samples were extracted from peripheral blood of DRS patients and unaffected family members. We performed exome sequencing on family members (III:11 and III:17; Fig. 1A). Exome sequencing and data analysis were conducted according to a previous study17 (Tables S1 and S2). Exonic variants—including missense variants, stop-codon variants, splice-site variants, and small frameshift and non-frameshift insertions and deletions (INDELs)—were filtered by ExAC, dbSNP, 1000 Genomes, and the ClinSeq database, based on a minor allele frequency below 1%. We also excluded synonymous variants within intergenic, intronic, and untranslated regions (UTRs). These variants were further filtered based on a heterozygous inheritance model. Moreover, the software packages, SIFT (https://sift.bii.astar.edu.sg/) and PolyPhen-2 (https://genetics.bwh.havard.edu/pph2), were used to evaluate potentially damaging effects of the variants on protein structure/function. Four online servers (I-Mutant 2.0, Site-Directed Mutator (SDM), mCSM and DUET)18,19,20,21, based on Gibbs free energy (indicated by a ΔΔG value), were used to assess the stability of each variant. The variations in ΔΔG were as follows: destabilizing (< 0 kcal/mol) and stabilizing (> 0 kcal/mol). The protein structure was predicted using I-TASSER. The PyMOL Molecular Graphics System (version 1.8; Schrödinger, LLC, New York City, NY, USA) was employed to determine functional changes in proteins in response to variants.
Sanger sequencing
CHN1 (RefSeq NM_001822.7) gene-variant analysis was performed by sequencing of the coding exons and the exon–intron boundaries of the CHN1 gene. Primers were designed according to the positions of coding exons, following the protocols of a previous study22. PCR amplification was performed in a 40-μL reaction mixture containing 50–100 ng of DNA, 2 mM of dNTP, 2 μM of each forward and reverse primer, 1 × LA Taq PCR buffer, and two units of LA Taq polymerase (TaKaRa, Japan). The PCR conditions were as follows: 95 °C for 5 min; 40 cycles of 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s; and a final extension of 72 °C for 5 min. Purified PCR products were directly sequenced using PCR primers and the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, CA, USA) on a 3730XL DNA sequencer (Applied Biosystems). DNASTAR SeqMan software (DNASTAR Inc, Madison, WI, USA) was used to analyze DNA sequences. Most of the family members and 200 unrelated control subjects from the same Southwestern Chinese population were sequenced to confirm gene variants. Newly determined sequences from the family members were deposited into GenBank (Accession numbers: MT919950-MT919974).
Functional analysis
Rac-GTP activation assay
Three constructs (V5 empty, V5 wild-type CHN1, and V5 variant p.(Phe213Val) CHN1) were constructed based on the pCDNA3.1 (+) vector and were transiently transfected into the human embryonic kidney (HEK) 293T cells. HEK293T cells were cultured at 37 °C in an incubator with 10% CO2. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, USA) supplemented with 10% fetal bovine serum (Gibco, USA), 1% glutamine, and 1% of a double-antibody. After 48 h in culture, a Rac-GTP activation assay was conducted using a Rac1-activation assay kit (Abcam, Cambridge, MA, USA), which was performed according to the manufacturer’s instructions. Briefly, cells were solubilized in lysis buffer. PAK1 PBD agarose beads were used to selectively pull-down active Rac from protein extracts. Subsequently, the precipitated GTP-Rac was detected via Western blotting using a mouse anti-Rac1 specific monoclonal antibody (BD Transduction Laboratories, San Jose, CA). β-actin was also detected—which was used as an internal reference—using β-actin mAB (Abmart, P3002, Arlington, MA, USA) and HRP-conjugated goat Anti-rabbit IgG (CST, 7074, USA). Secondary antibodies and signals were detected with an ECL detection system.
Quantification of α2-chimaerin translocation
After 48 h of transfection with wild-type and variant V5 constructs, HEK293T cells were pretreated with 5 μM of bisindolylmaleimide I (LC Laboratories, Woburn, MA) for 30 min to inhibit protein kinase C (PKC). Then, cells were divided into the following two groups (each group divided into six dishes): the first group was subjected to 10 μM of PMA (Sigma, Cambridge, MA, USA) stimulation for 20 min; and the other group was used as a control without stimulation. Next, three dishes of cells for each group were used for Western blotting. Other cells from each group were sonicated in CSK buffer23 and centrifuged at 100,000g at 4 °C for 1 h to separate the soluble and pelleted fractions (i.e., the membranous and cytoskeletal components). Each pellet was then resuspended in an equal volume of CSK buffer. Western blotting was performed with a goat polyclonal anti-V5 antibody (Abcam, ab9137, Cambridge, UK) and an HRP-conjugated rabbit Anti-goat IgG (Abcam, ab6885, USA).
Co-immunoprecipitation experiments
The GFP-fused wild-type_CHN1 vector was constructed base on the GV230 plasmid (GeneChem Co., Ltd., Shanghai, China). Specifically, 3 μg of V5 empty vector, V5 wild-type_CHN1 vector, or V5 variant_CHN1 vector was transiently co-transfected with 3 μg of GFP-fused wild-type _CHN1 vector into HEK293T cells. After 48 h of incubation, HEK293T cells were pretreated with 5 μM of a PKC inhibitor for 30 min and were then treated with 10 μM of PMA (Sigma, Cambridge, MA, USA) for 20 min. After 30 min of treatment on ice with lysis buffer, the cells were centrifuged at 18,000g at 4 °C for 20 min. The supernatant was precipitated using a rabbit polyclonal anti-GFP antibody (ab6556; Abcam, Cambridge, UK), and the pellet was then transferred to a nitrocellulose membrane. The signal was detected by a goat polyclonal anti-V5 antibody and an HRP-conjugated rabbit anti-goat IgG (Abcam, ab6885, USA). β-actin was also detected, as described in an earlier methods subsection, and was used as an internal control.
Statistical analysis
The grayscale results of Western blotting were used for semiquantitative analysis. Statistical differences were assessed with unpaired Student’s t-tests using GraphPad Prism 6 software. Differences were considered statistically significant at P values less than 0.05.
References
Duane, A. Congenital deficiency of abduction, associated with impairment of adduction, retraction movements, contraction of the palpebral fissure and oblique movements of the eye. Arch. Ophthalmol. (Chicago, Ill.:1960) 114, 1255–1256 (1905) (discussion 1257 (1996)).
DeRespinis, P. A., Caputo, A. R., Wagner, R. S. & Guo, S. Duane’s retraction syndrome. Surv. Ophthalmol. 38, 257–288 (1993).
Oystreck, D. T., Engle, E. C. & Bosley, T. M. Recent progress in understanding congenital cranial dysinnervation disorders. J. Neuroophthalmol. 31, 69–77. https://doi.org/10.1097/WNO.0b013e31820d0756 (2011).
Abu-Amero, K. K., Khan, A. O., Oystreck, D. T., Kondkar, A. A. & Bosley, T. M. The genetics of nonsyndromic bilateral Duane retraction syndrome. J. AAPOS 20, 396-400.e392. https://doi.org/10.1016/j.jaapos.2016.06.008 (2016).
Park, J. G. et al. Loss of MAFB function in humans and mice causes Duane syndrome, aberrant extraocular muscle innervation, and inner-ear defects. Am. J. Hum. Genet. 98, 1220–1227. https://doi.org/10.1016/j.ajhg.2016.03.023 (2016).
Miyake, N. et al. Human CHN1 mutations hyperactivate alpha2-chimaerin and cause Duane’s retraction syndrome. Science (New York, N.Y.) 321, 839–843. https://doi.org/10.1126/science.1156121 (2008).
Ip, J. P. et al. alpha2-chimaerin controls neuronal migration and functioning of the cerebral cortex through CRMP-2. Nat. Neurosci. 15, 39–47. https://doi.org/10.1038/nn.2972 (2011).
Beg, A. A., Sommer, J. E., Martin, J. H. & Scheiffele, P. alpha2-Chimaerin is an essential EphA4 effector in the assembly of neuronal locomotor circuits. Neuron 55, 768–778. https://doi.org/10.1016/j.neuron.2007.07.036 (2007).
Colon-Gonzalez, F., Leskow, F. C. & Kazanietz, M. G. Identification of an autoinhibitory mechanism that restricts C1 domain-mediated activation of the Rac-GAP alpha2-chimaerin. J. Biol. Chem. 283, 35247–35257. https://doi.org/10.1074/jbc.M806264200 (2008).
Iwasato, T. et al. Rac-GAP alpha-chimerin regulates motor-circuit formation as a key mediator of EphrinB3/EphA4 forward signaling. Cell 130, 742–753. https://doi.org/10.1016/j.cell.2007.07.022 (2007).
Appukuttan, B. et al. Localization of a gene for Duane retraction syndrome to chromosome 2q31. Am. J. Hum. Genet. 65, 1639–1646. https://doi.org/10.1086/302656 (1999).
Al-Baradie, R. et al. Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and results from mutations in SALL4, a new member of the SAL family. Am. J. Hum. Genet. 71, 1195–1199. https://doi.org/10.1086/343821 (2002).
Chung, M., Stout, J. T. & Borchert, M. S. Clinical diversity of hereditary Duane’s retraction syndrome. Ophthalmology 107, 500–503 (2000).
Calabrese, G. et al. Narrowing the Duane syndrome critical region at chromosome 8q13 down to 40 kb. EJHG 8, 319–324. https://doi.org/10.1038/sj.ejhg.5200461 (2000).
Miyake, N. et al. Expansion of the CHN1 strabismus phenotype. Invest. Ophthalmol. Vis. Sci. 52, 6321–6328. https://doi.org/10.1167/iovs.11-7950 (2011).
Chan, W. M., Miyake, N., Zhu-Tam, L., Andrews, C. & Engle, E. C. Two novel CHN1 mutations in 2 families with Duane retraction syndrome. Arch. Ophthalmol. (Chicago, Ill.:1960) 129, 649–652. https://doi.org/10.1001/archophthalmol.2011.84 (2011).
Calender, A. et al. Whole exome sequencing in three families segregating a pediatric case of sarcoidosis. BMC Med. Genom. 11, 23. https://doi.org/10.1186/s12920-018-0338-x (2018).
Capriotti, E., Fariselli, P., Calabrese, R. & Casadio, R. Predicting protein stability changes from sequences using support vector machines. Bioinformatics (Oxford, England) 21(Suppl 2), ii54–ii58. https://doi.org/10.1093/bioinformatics/bti1109 (2005).
Pires, D. E., Ascher, D. B. & Blundell, T. L. mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics (Oxford, England) 30, 335–342. https://doi.org/10.1093/bioinformatics/btt691 (2014).
Pandurangan, A. P., Ochoa-Montano, B., Ascher, D. B. & Blundell, T. L. SDM: a server for predicting effects of mutations on protein stability. Nucleic Acids Res. 45, W229-w235. https://doi.org/10.1093/nar/gkx439 (2017).
Pires, D. E., Ascher, D. B. & Blundell, T. L. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 42, W314-319. https://doi.org/10.1093/nar/gku411 (2014).
Biler, E. D., Ilim, O., Onay, H. & Uretmen, O. CHN1 gene mutation analysis in patients with Duane retraction syndrome. J. AAPOS 21, 472-475.e472. https://doi.org/10.1016/j.jaapos.2017.07.208 (2017).
Kozma, R., Ahmed, S., Best, A. & Lim, L. The GTPase-activating protein n-chimaerin cooperates with Rac1 and Cdc42Hs to induce the formation of lamellipodia and filopodia. Mol. Cell. Biol. 16, 5069–5080. https://doi.org/10.1128/mcb.16.9.5069 (1996).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81560168 and 81560169), Reserve Talents of Young and Middle-aged Academic and Technical Leaders of Yunnan Province (2018HB048), Yunnan Provincial Department of Science and Technology- Kunming Medical University Joint Fund 2019FE001(-095), and Expert Workstation of Yao Ke (2017IC064).
Author information
Authors and Affiliations
Contributions
All authors contributed to and have approved the final manuscript. T.C.Z., M.H., W.H.D., and X.L.F. designed the study; L.Y.G., Q.Z., and Y.Z. collected the samples and clinical information; X.F.Z., Z.J.H., X.J.L., and D.M.Y. performed the experimental procedures; J.Y.Z., J.Y., and G.L.Z. performed the data analysis; X.L.F., T.C.Z., and W.H.D. drafted the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, TC., Duan, WH., Fu, XL. et al. Identification of a novel CHN1 p.(Phe213Val) variant in a large Han Chinese family with congenital Duane retraction syndrome. Sci Rep 10, 16225 (2020). https://doi.org/10.1038/s41598-020-73190-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-73190-1
- Springer Nature Limited
This article is cited by
-
Two novel CHN1 variants identified in Duane retraction syndrome pedigrees disrupt development of ocular motor nerves in zebrafish
Journal of Human Genetics (2024)