
Abstract
Birgus latro (coconut crab) is an edible crustacean that has experienced serious overharvesting throughout its whole habitat range; however, the negative effects of overharvesting on the genetic diversity within B. latro populations have not been elucidated. Here, we report sex ratio, body size, and genetic diversity in populations of B. latro in the Ryukyu Islands where large-male–biased overharvesting of B. latro has continued. In 2 of the study populations, the sex ratio was significantly skewed toward females, and in all of the study populations large males were rare, which we attributed to sex- and size-biased overharvesting. We found no differences in genetic diversity between small and large individuals, suggesting that genetic diversity, even among the large (i.e., old) individuals, may have had already been negatively affected by overharvesting. Continued monitoring of sex ratio, body size and genetic diversity are needed for effective management of the study populations.
Similar content being viewed by others


Introduction
Overharvesting drives loss of genetic diversity1,2,3. Once genetic diversity is lost from a population, it can be restored by genetic mutation or immigration of individuals from a population with high genetic diversity. However, the recovery of genetic diversity through mutation takes a long time, and although immigration from refugia is faster than mutation, the restoration of genetic diversity in isolated populations cannot be expected to occur through this means3. Therefore, conserving genetic diversity and increasing our understanding of gene flow patterns should be important goals for the effective management of fishery resources3,4,5.
Individual genetic diversity (individual heterozygosity) likely plays an important role in population sustainability because it is correlated with fitness. For example, a meta-analysis has shown that the correlation between the level of individual heterozygosity and fitness is small but significantly positive6, although outbreeding depression has been shown to cause the correlation to become negative7. In addition, many papers have reported significant positive relationships between individual heterozygosity of allozyme or neutral microsatellite markers and fitness-related traits in taxa such as fish8,9,10,11,12, mollusks13,14, crustaceans15, marine and terrestrial mammals16,17,18,19, and terrestrial plants20. It is therefore important to investigate genetic diversity to assess the sustainability of fishery resources.
High, long-term harvesting pressure often causes the average body size within a population to decrease21. Body size is a key factor that influences fitness in many taxa22,23. In males, larger individuals are stronger competitors for females24 and provide more sperm per ejaculation25, and in females, body size is positively correlated with number of eggs26, egg size27, and larval body size and starvation resistance28.
The coconut crab (Birgus latro) is a terrestrial hermit crab with a marine larval dispersal stage distributed in subtropical and tropical regions of the Indo-Pacific29,30. Recent report showed that this species predates other animals including birds and mammals, and can have strong impacts on prey behavior, abundance and community composition31. However, this species has suffered severe resource depletion throughout its entire distribution range due to overharvesting and habitat destruction30,31,32,33,34,35. Therefore, B. latro is currently categorized as “data deficient” in the International Union for Conservation of Nature’s Red List, and “vulnerable” in the Japanese Ministry of the Environment’s Red Data Book36. In Japan, B. latro is an important resource not only for food but also for culture and tourism37. However, selective harvesting of large males has resulted in the sex ratio of local populations becoming skewed toward females and the average male body size becoming miniaturized in places that have experienced high fishery pressure38. Recent studies have revealed that large-male–biased harvesting negatively affects populations in several ways39: it decreases male size-dependent reproductive potential [e.g., number of retained sperm40, number of possible mates41, and number of ejaculated sperm42], reduces the pool of suitable males due to females refusing to mate with males smaller than themselves38, which results in reductions of number of spawned eggs43 and larval qualities44]. Thus, it is likely that decreases of average body size within a population will have negative impacts on the population’s genetic diversity through massive decrease of effective population size.
Recently, the use of multiplexed inter-simple sequence repeat (ISSR) genotyping by sequencing (MIG-seq)45 to analyze large numbers of genome-wide markers has become a useful tool for examining the genetic structure and diversity of populations at high temporal and spatial resolution46. Here, we examined sex ratio, thoracic length (as an index of body size), and genetic diversity (by COI gene and MIG-seq analyses) to elucidate the effects of overharvesting on population structure and genetic diversity as well as gene flow pattern among 8 populations (Fig. 1) of B. latro in the Ryukyu Islands, Japan.

Map showing sampling localities of B. latro in the Ryukyu Islands. (a) A map of east Asia. (b) A map of sampling location of the present study in the Ryukyu Islands, Japan.
Results
Sex ratio and body size
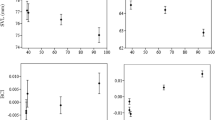
In all study populations except Kurima (MK), the number of females was higher than that of males, although the difference was only significant in Ishigaki (IS) and Hatoma (HT) (Table 1). Female thoracic length in MK was significantly smaller than that in IE, TM, IS and HT. Male thoracic length in MK was significantly smaller than that in MY, TM, HT and YG (Fig. 2b; Table S1). In addition, male thoracic length in TM was significantly larger than that in IE, MK, IS, HT and YG (Fig. 2b; Table S1).

Box plot showing thoracic length of female (a) and male (b) B. latro individuals in each population.
Correlation test with Spearman’s rank correlation coefficient showed no significant correlation between cumulative human population density and, body size and proportion of female individuals (Fig. S1).
Genetic diversity and population genetic structure
COI gene analysis
We sequenced a 466-bp region of the mtDNA COI gene sequence in 154 individuals and detected 53 haplotypes. Four dominant haplotypes were found in most of the study populations (Fig. 3a). Population pairwise PhiPT was low overall (PhiPT < 0.018), and no significant differences were found among the populations (Table S2). MK and TM showed low genetic diversity compared with the other populations (Table S3). No marked difference of genetic diversity was detected between small and large individuals for either sex (Fig. 3b; Table S3).

Haplotype network of B. latro showing frequency of mtDNA COI haplotypes in each population (a) and size class of both sexes (b). FL, large female; ML, large male, FS, small female; MS, small male.
Correlation test with Spearman’s rank correlation coefficient showed no significant correlation between cumulative human population density and, haplotype diversity and nucleotide diversity, (Fig. S1).
MIG-seq analysis
We used 495 neutral genetic markers for the genetic analysis. TM and IS were significantly differentiated from both MK and YG (FST = 0.074–0.104, P < 0.01; Table S2). No significant relationship between genetic distance and geographic distance was detected by Mantel’s test (R2 = 0.042, P = 0.232; Fig. 4). Observed heterozygosity was low in TM (0.041) and I (0.042), and high in M (0.059), compared with that in the other populations (Table S3). Fixation index values were high in all populations (F = 0.294–0.532; Table S3). The directional relative migration network for the study populations indicated that IE, MK, HT, IR, and YG are core populations that have high gene flows among each other, whereas MY, TM, and IS are peripheral populations with low gene flow from other populations (Fig. 5). In particular, immigration to TM and IS was suggested to be very limited (Fig. 5b). No significant asymmetric pattern of gene flow was detected. In both sexes, individual heterozygosity did not show positive correlation with body size (Fig. 6).

Relationship between pairwise population genetic distance inferred from MIG-seq SNP markers and geographical distance in Birgus latro.

Directional relative migration networks of B. latro populations constructed with divMigrate using Nm. Values above 0.2 (a) and 0.4 (b) are shown.

Plot showing body size and mean observed individual heterozygosity of female (a) and male (b) B. latro. Lines indicate linear regression.
Correlation test with Spearman’s rank correlation coefficient showed no significant correlation between cumulative human population density and observed heterozygosity (Fig. S1).
Discussion
Body size and sex ratio
In Japan, large males of B. latro are selectively harvested, which has caused the sex ratio to become skewed toward females and average male body size to become miniaturized in heavily harvested populations38. In the present study, sex ratio was significantly skewed toward females in populations IS and HT, suggesting that male-selective harvesting pressure is higher in IS and HT than in the other populations. However, past human population density was not correlated with body size, sex ratio and genetic diversities of B. latro (Fig. S1), suggesting that human population density does not simply reflect the intensity of fishery pressure on B. latro. Although adult male and female B. latro are expected to reach a thoracic length of 80 mm and 60 mm, respectively29, in the present study large males (>40 mm) were very rarely seen except for TM, even in the populations where the sex ratio was less, or not, skewed toward females. A lack of large males promotes resource reduction through negative impacts on life history parameters (e.g., decrease of mating opportunity)39. Our findings suggest that urgent implementation of an effective management policy is needed in Ryukyu Islands.
Genetic diversity and gene flow
Birgus latro can live for 60 years47 or longer48. At 10 years of age, males and females generally have a thoracic length of around 30 mm and 25 mm, respectively49. Overharvesting of B. latro likely started in Japan after the end of the Second World War37, and although the need for conservation efforts was noted as early as the 1970s, we were told by residents of Okinawa that overharvesting has continued and even become more severe in the past two decades (Fujita, personal communication). Thus, most small individuals analysed in the present study (male, thoracic length <30 mm; female, <25 mm) would have been recruited after the intensification of overharvesting, whereas some of the largest individuals could have been recruited before. This suggests that since overharvesting reduces genetic diversity within a population1,2,3, the genetic diversity of small B. latro individuals should be lower than that of large individuals. However, in the present study, we did not find a correlation between thoracic length and population- or individual-level genetic diversity, as estimated by using COI gene and MIG-seq data. The average thoracic length of large individuals used in the COI gene analysis was 40 mm and 25 mm for males and females, respectively; therefore, based on the growth curve of B. latro, most of these individuals were recruited in the last 15–20 years49. The thoracic length of most individuals used for the MIG-seq analysis was smaller than 40 mm for both sexes; therefore, based on the growth curve of B. latro, most of these individuals were recruited within the last 20 years for males and within the last 30 years for females. It is therefore, suggests that not a few individuals included in the genetic analyses were likely recruited after the intensification of overharvesting and may have already suffered the impacts of overharvesting on genetic diversity. Growth rate can, however, vary among individuals based on resource availability and levels of social competition (e.g., growth can be slowed considerably when individuals lose a limb in combat). Further monitoring and analysis using more samples from each population with wide range of body size are needed to evaluate the effect of overharvesting on genetic diversity of B. latro.
To conserve genetic diversity, genetic mutation or immigration from other populations with high genetic diversity are needed3. Once genetic diversity is lost, recovery by genetic mutation takes many generations, and as both the coastal and oceanic environments are being rapidly changed by human activity50,51,52, mutation-based recovery is not realistic. In contrast, immigration-based recovery can occur relatively quickly3. In the COI analysis, no significant genetic differentiation between the study populations was found, and in the MIG-seq analysis, most population pairs showed no significant genetic differentiation. The pelagic zoeal larval stage of B. latro lasts for 18–23 days53,54, and megalopae settle at coastal areas and find a gastropod shell for migrating to land by around 10 days post settlement55,56. Therefore, the whole larval period lasts 4–5 weeks, which is similar to that of the coral Acropora digitifera57. It has been reported that population connectivity is generally high among A. digitifera populations in the Ryukyu Islands58, and the sampling locations in that study were similar to those in the present study. This suggests that the larval period of B. latro is long enough to allow sufficient larval dispersal among the study populations. This assumption is supported by the results of the Mantel test, which showed a non-significant correlation between genetic distance and geographic distance, which suggests that migration via larval dispersal frequently occurs among Japanese populations of B. latro.
Although a high gene flow has been maintained in B. latro populations in the Ryukyu Islands, MIG-seq analysis detected significant genetic differentiation between four population pairs: TM–MK, TM–YG, IS–MK, and IS–YG. This is consistent with the divMigrate results showing that immigration to TM and IS from other populations is limited and IE, MK, HT, YG, and IR have a role as core populations within the overall population in the Ryukyu Islands. Local water current can work as a dispersal barrier in Ryukyu islands59. In addition, TM is far from the large islands (Fig. 1). These factors may contribute to the limitation of immigration to TM and IS. Similarly, genetic differentiation has been shown between populations in the Indian and Pacific Oceans60,61 and between populations in the Ryukyu Islands and populations in Micronesia, Palau, and Indonesia62, indicating that immigration from the Indian Ocean, Micronesia, Palau, and Indonesia to the Ryukyu Islands is likely limited. All populations analyzed in the present study had high fixation index values, suggesting high rates of inbreeding and that a further reduction of effective population size driven by overharvesting and habitat degradation may reduce gene flows and drastically increase the risk of inbreeding depression.
Here, we report COI- and MIG-seq-based genetic diversity, sex ratio and body size distributions in 8 Japanese populations of B. latro. We found that 5 populations (IE, MK, HT, YG, and IR) that are important for the maintenance of the genetic diversity of the other populations via immigration through larval dispersal. Low genetic diversity can affect the fitness6 and ability of individuals to survive and adapt in future environments3. To conserve the genetic diversity of B. latro populations in Japan, and therefore to conserve B. latro as an important fishery resource, we recommend long-term monitoring of genetic diversity, sex ratio, and body size composition. In Okinawa prefecture (the administrative division that includes the Ryukyu Islands), several municipal governments have implemented regulations that prohibit the catch of B. latro of certain sizes; we hope that such regulation will be implemented soon across the whole region.
Methods
Field surveys and sample collection
Field surveys were conducted in Ie (IE), Miyako (MY), Kurima (MK), Minna (TM), Ishigaki (IS), Hatoma (HT), Iriomote (IR), and Yonaguni (YG) in the Ryukyu Islands, Japan (Fig. 1; Table 1). Individuals of B. latro were randomly collected by hand and the sex and thoracic length (as an index of body size) of each individual were recorded. For DNA analysis, part of the third pereiopod was also collected. Tissue samples were fixed and preserved in 99.5% ethanol. All crabs were released at the sampling site after tissue sampling. Field surveys and sample collection were conducted in accordance with the regulations of local governments in Okinawa Prefecture, Japan.
Statistical analyses
We examined whether the sex ratio of each population is skewed toward either sex by using the χ2-test. In addition, we compared thoracic length among populations for each sex by using Tukey’s test. All analyses were performed using R v3.3.063.
We assumed that the past human population density reflects the intensity of fishery pressure on B. latro. We therefore tested correlation between cumulative human population densities and, sex ratio, median body size, and genetic diversity indexes of B. latro in each island population with Spearman’s rank correlation coefficient. Data of every 5 year human population densities (1955–2015) of each island was obtained from Okinawa Prefecture64. We cumulated the human population density of 1955–2015 (Table 1), and used for the analyses.
mtDNA COI gene sequencing
DNA was extracted from tissue samples by using a DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany). Partial sequences of the mtDNA COI-encoding region were amplified by polymerase chain reaction (PCR) with the universal primers LCO1490 and HCO219865, as well as TaKaRa Ex Taq (TaKaRa, Shiga, Japan). The PCR conditions were as follows: initial denaturation at 94 °C for 2 min; 35 cycles each at 94 °C for 30 s, annealing at 47 °C for 30 s, and extension at 72 °C for 1 min; and a final extension step at 72 °C for 2 min. The PCR products were purified using Exo-SAP IT (Affymetrix, USB, Cleveland, USA) and sequenced using an Applied Biosystems 3730xl DNA Analyser and the same primers as used for the PCR.
MIG-seq
Genome-wide single nucleotide polymorphisms (SNPs) were obtained using the protocol45. In brief, MIG-seq was used to amplify a few hundred to a few thousand genome-wide SNPs around ISSRs by using eight universal pairs of multiplex ISSR primers (MIG-seq primer set 1) for the first PCR. Then, DNA libraries with different indexes were pooled and sequenced by using a MiSeq system (sequencing control software v2.0.12, Illumina) and a MiSeq Reagent Kit v3 (150 cycle) (Illumina). A total of 83 individuals were analyzed in the MIG-seq analysis.
To eliminate low-quality reads and primer sequence reads from the raw data, we used the FASTX-Toolkit v0.0.14 (fastq_quality_filter) (http://hannonlab.cshl.edu/fastx_toolkit/index.html) with a fastq-quality-filter setting of –Q 33 –q 30 –p 40. We removed adapter sequences for the MiSeq run from both the 5′ end (GTCAGATCGGAAGAGCACACGTCTGAACTCCAGTCAC) and 3′ end (CAGAGATCGGAAGAGCGTCGTGTAGGGAAAGAC) by using Cutadapt v1.1366, and then excluded short reads less than 80 bp. The quality-filtered sequence data were demultiplexed and filtered through the software Stacks v1.4667,68. We used Stacks v1.468 to stack the reads and extract SNPs. First, we used the U-stacks program with the following settings: ‘minimum depth of coverage required to create a stack (m)’ = 3, ‘maximum distance allowed between stacks (M)’ = 1, ‘maximum distance allowed to align secondary reads to primary stacks (N)’ = 1, with the deleveraging and removal algorithms enabled. Then, we used the C-stacks program with the option ‘number of mismatches allowed between sample loci when building the catalog (n)’ = 4, followed by the S-stacks program. Finally, we used the Populations program in Stacks v1.4 by restricting the data analysis to the criteria (the minimum percentage of individuals required to process a locus across all data was set at 50% and restricting the data analysis to a single SNP per locus. No locus was identified as an outlier using BayScan69.
Population genetic analyses
mtDNA COI gene analysis
Haplotype networks were constructed using the haploNet function in the R package ‘pegas’ v0.1170. Haplotype diversity and nucleotide diversity were calculated for each population by using Arlequin 3.571. In addition, we pooled all sequence data for each sex and divided them into two size groups, small and large, with the small group including males with thoracic length < 30 mm and females with thoracic length < 25 mm (i.e., individuals roughly less than 10 years old)49, and calculated genetic diversities for the two size classes. Mean ± standard deviation of the thoracic length of each group is 23.6 ± 4.0 mm (N = 25), 35.1 ± 5.6 mm (N = 29), 23.2 ± 2.5 mm (N = 22), and 31.6 ± 3.9 mm (N = 62) for small male, large male, small female, and large female, respectively. Population pairwise PhiPT values were estimated using the Analysis of Molecular Variance (AMOVA) method and GenAlEx 6.572 and tested for significance based on 999 permutations. Statistical significance levels for all pairwise tests were 0.05 after adjusting for multiple comparisons using false discovery rate (FDR) correction73.
MIG-seq
Based on 495 SNP markers, ratio of the number of observed alleles, observed heterozygosity, expected heterozygosity, and fixation index were estimated using GenAlEx 6.572. Pairwise population FST values were estimated by using the AMOVA method and GenAlEx 6.5. Statistical significance levels for all pairwise tests were 0.05 after adjusting for multiple comparisons using FDR correction68. Isolation by distance was tested using Mantel’s test based on 9999 permutations from the comparison of all pairwise FST/(1 − FST) values with pairwise geographic distances in kilometers (straight-line distance) using GenAlEx 6.572. In addition, individual heterozygosity was estimated using GENHET v2.374 with R v3.3.063.
The gene flow among populations was estimated by using divMigrate-online75 (https://popgen.shinyapps.io/divMigrate-online/). This program produces a migration network graph with relative values for gene flow among populations scaled to the largest magnitude estimated. For the analysis, we selected 61 MIG-seq markers that were detected in at least 68% of the samples. We used NM as a measure of genetic distance. The significance of asymmetrical gene flow among populations was tested using 1000 bootstrap iterations.
Data availability
Raw data of body length and sex ratio of B. latro are available in supplementary information. mtDNA COI sequences (Accession nos. LC479132- LC479285) and law data of MIG-seq (Accession no. PRJDB8390) were deposited in DNA Data Bank of Japan (DDBJ).
References
Ryman, N., Utter, F. & Laikre, L. Protection of intraspecific biodiversity of exploited fishes. Rev. Fish Biol. Fisheries. 5, 417–446 (1995).
Allendorf, F. W., England, P. R., Luikart, G., Ritchie, P. A. & Ryman, N. Genetic effects of harvest on wild animal populations. Trends Ecol. Evol. 23, 327–337 (2008).
Pinsky, M. L. & Palumbi, S. R. Meta-analysis reveals lower genetic diversity in overfished populations. Mol. Ecol. 23, 29–39 (2014).
Smith, P. J. Genetic diversity of marine fisheries resources: possible impacts of fishing. FAO Fisheries Technical Paper 344, 1–53 (1994).
Jorgensen, C. et al. Ecology-Managing evolving fish stocks. Science 318, 1247–1248 (2007).
Chapman, J. R., Nakagawa, S., Coltman, D. W., Slate, J. & Sheldon, B. C. A quantitative review of heterozygosity–fitness correlations in animal populations. Mol. Ecol. 18, 2746–2765 (2009).
Szulkin, M. & David, P. Negative heterozygosity–fitness correlations observed with microsatellites located in functional areas of the genome. Mol. Ecol. 20, 3949–3952 (2011).
Danzmann, R. G., Ferguson, M. M. & Allendorf, F. W. Heterozygosity and oxygen-consumption rate as predictors of growth and developmental rate in rainbow trout. Physiol. Zool. 60, 211–220 (1987).
Thelen, G. C. & Allendorf, F. W. Heterozygosity-fitness correlations in rainbow trout: effects of allozyme loci or associative overdominance? Evolution 55, 1180–1187 (2001).
Pujolar, J. M., Maes, G. E., Vancoillie, C. & Volckaert, F. A. M. Growth rate correlates to individual heterozygosity in the European eel. Anguilla anguilla L. Evolution 59, 189–199 (2005).
Fassatoui, C., Chenuil, A. & Romdhane, M. S. Relationships between heterozygosity, growth parameters and age in the common pandora Pagellus erythrinus (Sparidae) in the Gabes Gulf (Tunisia). Mar. Ecol. Prog. Ser. 445, 251–261 (2012).
Guinand, B. et al. Genetic structure and heterozygosity–fitness correlation in young-of-the-year sole (Solea solea L.) inhabiting three contaminated West-European estuaries. J. Sea Res. 80, 35–49 (2013).
Zouros, E., Singh, S. M. & Miles, H. E. Growth rate in oysters: an overdominant phenotype and its possible explanations. Evolution 34, 856–867 (1980).
Koehn, R. K. & Gaffney, P. M. Genetic heterozygosity and growth rate in Mytilus edulis. Mar. Biol. 82, 1–7 (1984).
Bierne, N., Beuzart, I., Vonau, V., Bonhomme, F. & Bédier, E. Microsatellite-associated heterosis in hatchery-propagated stocks of the shrimp Penaeus stylirostris. Aquaculture 184, 203–219 (2000).
Coltman, D. W., Bowen, W. D. & Wright, J. M. Birth weight and neonatal survival of harbour seal pups are positively correlated with genetic variation measured by microsatellites. Proc. R. Soc. Lond. B Biol. Sci. 265, 803–809 (1998).
Coulson, T. N. et al. Microsatellites reveal heterosis in red deer. Proc. R. Soc. Lond. B Biol. Sci. 265, 489–495 (1998).
Coltman, D. W., Pilkington, J. G., Smith, J. A. & Pemberton, J. M. Parasite-mediated selection against inbred soay sheep in a free-living island population. Evolution 53, 1259–1267 (1999).
Slate, J. & Pemberton, J. M. Comparing molecular measures for detecting inbreeding depression. J. Evol. Biol. 15, 20–31 (2002).
Ledig, F. T., Guries, R. P. & Bonefeld, B. A. The relation of growth to heterozygosity in pitch pine. Evolution 37, 1227–1238 (1983).
Jackson, J. B. et al. Historical overfishing and the recent collapse of coastal ecosystems. Science 293, 629–637 (2001).
Blueweiss, L. et al. Relationships between body size and some life history parameters. Oecologia 37, 257–272 (1978).
Anderson, M. Sexual Selection. (Princeton University Press,1994).
Jormalainen, V. Precopulatory mate guarding in crustaceans: male competitive strategy and intersexual conflict. Q. REV. Biol. 73, 275–304 (1998).
Sato, T., Ashidate, M., Jinbo, T. & Goshima, S. Variation of sperm allocation with male size and recovery rate of sperm numbers in spiny king crab Paralithodes brevipes. Mar. Ecol. Prog. Ser. 312, 189–199 (2006).
Sato, T. & Goshima, S. Impacts of male-only fishing and sperm limitation in manipulated populations of an unfished crab, Hapalogaster dentata. Mar. Ecol. Prog. Ser. 313, 193–204 (2006).
Moland, E., Moland, O. E. & Stenseth, N. C, Maternal influences on offspring size variation and viability in wild European lobster Homarus gammarus. Mar. Ecol. Prog. Ser. 400, 165–173 (2010).
Sato, T., Hamano, K., Sugaya, T. & Dan, S. Effects of maternal influences and timing of spawning on intraspecific variations in larval qualities of the Kuruma prawn Marsupenaeus japonicus. Mar. Biol. 164, 70 (2017).
Drew, M. M., Harzsch, S., Stensmyr, M., Erland, S. & Hansson, B. S. A review of the biology and ecology of the robber crab, Birgus latro (Linnaeus, 1767) (Anomura: Coenobitidae). Zool. Anz. 249, 45–67 (2010).
Laidre, M. E. Coconut crabs. Curr. Biol. 28, 58–60 (2018).
Laidre, M. E. Ruler of the atoll: the world’s largest land invertebrate. Front. Ecol. Environ. 15, 527–528 (2017).
Amesbury, S. S. Biological studies on the coconut crab (Birgus latro) in the Mariana Islands. (Agricultural Experiment Station, College of Agriculture and Life Sciences, University of Guam, 1980).
Brown, I. W., & Fielder, D. R. Project overview and literature survey in The Coconut Crab: Aspects of Birgus latro Biology and Ecology in Vanuatu, ACIAR Monograph 8 (eds. Brown, I.W. & Fielder, D. R.) 1–11 (Australian Centre for International Agricultural Research, 1991).
Fletcher, W. J. Coconut crabs in Nearshore Marine Resources of the South Pacific (eds. Wright, A. & Hill, L.) 643–681 (Institute of Pacific Studies, University of the South Pacific, 1993).
Eldredge, L. G. Birgus latro. IUCN Red List of Threatened Species (1996). Available at: http://www.iucnredlist.org/details/2811/0. (Accessed 9 January 2019).
Ministry of the Environment, Government of Japan. The 4th Version of the Red Data Book (2018). Website https://ikilog.biodic.go.jp/Rdb/booklist (accessed 27 February 2019) (in Japanese).
Fujita, Y. Yashigani to hitobito no kurashi. Cancer 19, 41–51 (2010). in Japanese.
Sato, T. & Yoseda, K. Influence of size-and sex-biased harvesting on reproduction of the coconut crab Birgus latro. Mar. Ecol. Prog. Ser. 402, 171–178 (2010).
Sato, T. Impacts of large male-selective harvesting on reproduction: illustration with large decapod crustacean resources. Aqua-BioSci. Monogr. 5, 67–102 (2012).
Sato, T., Yoseda, K., Abe, O. & Shibuno, T. Male maturity, number of sperm, and spermatophore size relationships in the coconut crab Birgus latro on Hatoma Island, southern Japan. J. Crust. Biol. 28, 663–668 (2008).
Sato, T. Plausible causes for sperm-store variations in the coconut crab Birgus latro under large male-selective harvesting. Aquat. Biol. 13, 11–19 (2011).
Sato, T., Yoseda, K., Okuzawa, K. & Suzuki, N. Sperm limitation: possible impacts of large male-selective harvesting on reproduction of the coconut crab Birgus latro. Aquat. Biol. 10, 23–32 (2010).
Sato, T. & Yoseda, K. Reproductive season and female maturity size of coconut crab Birgus latro in Hatoma Island, southern part of Japan. Fish. Sci. 74, 1277–1282 (2008).
Sato, T. & Suzuki, N. Female size as a determinant of larval size, weight, and survival period in the coconut crab, Birgus latro. J. Crust. Biol. 30, 624–628 (2010).
Suyama, Y. & Matsuki, Y. MIG-seq: an effective PCR-based method for genome-wide single-nucleotide polymorphism genotyping using the next-generation sequencing platform. Sci. Rep. 5, 16963 nature.com/articles/srep16963 (2015).
Wachi, N., Matsubayashi, K. W. & Maeto, K. Application of next-generation sequencing to the study of non-model insects. Entomol. Sci. 21, 3–11 (2018).
Fletcher, W. J., Brown, I. W. & Fielder, D. R. Moulting and growth characteristics in The Coconut Crab: Aspects of Birgus latro Biology and Ecology in Vanuatu, ACIAR Monograph 8 (eds. Brown, I.W. & Fielder, D. R.) 35–60 (Australian Centre for International Agricultural Research, 1991).
Drew, M. M., Smith, M. J. & Hansson, B. S. Factors influencing growth of giant terrestrial robber crab Birgus latro (Anomura: Coenobitidae) on Christmas Island. Aquat. Biol. 19, 129–141 (2013).
Sato, T. et al. Growth of the coconut crab Birgus latro estimated from mark-recapture using passive integrated transponder (PIT) tags. Aquat. Biol. 19, 143–152 (2013).
Swartz, W., Sala, E., Tracey, S., Watson, R. & Pauly, D. The spatial expansion and ecological footprint of fisheries (1950 to present). PloS One 5(12), e15143, https://doi.org/10.1371/journal.pone.0015143 (2010).
McCauley, D. J. et al. Marine defaunation: animal loss in the global ocean. Science 347, 1255641 (2015).
Cloern, J. E. et al. Human activities and climate variability drive fast-paced change across the world’s estuarine–coastal ecosystems. Global Change Biol. 22, 513–529 (2016).
Hamasaki, K., Sugizaki, M., Dan, S. & Kitada, S. Effect of temperature on survival and developmental period of coconut crab (Birgus latro) larvae reared in the laboratory. Aquaculture 292, 259–263 (2009).
Hamasaki, K., Kato, S., Murakami, Y., Dan, S. & Kitada, S. Larval growth, development and duration in terrestrial hermit crabs. Sex. Early Dev. Aquat. Org. 1, 93–107 (2015).
Hamasaki, K., Sugizaki, M., Sugimoto, A., Murakami, Y. & Kitada, S. Emigration behaviour during sea-to-land transition of the coconut crab Birgus latro: effects of gastropod shells, substrata, shelters and humidity. J. Exp. Mar. Biol. Ecol. 403, 81–89 (2011).
Hamasaki, K., Ishiyama, N. & Kitada, S. Settlement behavior and substrate preference of the coconut crab Birgus latro megalopae on natural substrata in the laboratory. J. Exp. Mar. Biol. Ecol. 468, 21–28 (2015).
Nishikawa, A. & Sakai, K. Settlement-competency period of planulae and genetic differentiation of the scleractinian coral Acropora digitifera. Zool. Sci. 22, 391–399 (2005).
Nakajima, Y., Nishikawa, A., Iguchi, A. & Sakai, K. Gene flow and genetic diversity of a broadcast-spawning coral in northern peripheral populations. PLoS One 5, e11149, https://doi.org/10.1371/journal.pone.0011149 (2010).
Weese, D. A., Fujita, Y., Hidaka, M. & Santos, S. R. The long and short of it: Genetic variation and population structure of the anchialine atyid shrimp Caridina rubella on Miyako-Jima, Japan. J. Crust. Biol. 32, 109–117 (2012).
Lavery, S., Moritz, C. & Fielder, D. R. Changing patterns of population structure and gene flow at different spatial scales in Birgus latro (the coconut crab). Heredity 74, 531 (1995).
Lavery, S., Moritz, C. & Fielder, D. R. Indo-Pacific population structure and evolutionary history of the coconut crab Birgus latro. Mol. Ecol. 5, 557–570 (1996).
Hamasaki, K. et al. Genetic diversity and demographic history of the terrestrial hermit crabs Birgus latro and Coenobita brevimanus in the north-western Pacific region. J. Crust. Biol. 35, 793–803 (2015).
R core team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria (2016).
Okinawa Prefecture. Riyou kankei siryou. (2017). Website https://www.pref.okinawa.jp/site/kikaku/chiikirito/ritoshinko/h28ritoukankeisiryou.html (accessed 5 December 2019) (in Japanese).
Folmer, O., Black, M., Hoeh, W., Lutz, R. & Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnology 3, 294–299 (1994).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal 1, 10–12 (2011). embnetjournal/article/view/200.
Catchen, J. M., Amores, A., Hohenlohe, P., Cresko, W. & Postlethwait, J. H. Stacks: building and genotyping loci de novo from short-read sequences. G3. 1, 171–182 (2011).
Catchen, J., Hohenlohe, P. A., Bassham, S., Amores, A. & Cresko, W. A. Stacks: an analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140 (2013).
Foll, M. & Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180, 977–993 (2008).
Paradis, E. pegas: an R package for population genetics with an integrated–modular approach. Bioinformatics 26, 419–420 (2010).
Excoffier, L. & Lischer, H. E. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Res. 10, 564–567 (2010).
Peakall, P. E. & Smouse, R. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28, 2537–2539 (2012).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Royal Stat. Soc. B 57, 289–300 (1995).
Coulon, A. GENHET: an easy-to-use R function to estimate individual heterozygosity. Mol. Ecol. Res. 10, 167–169 (2010).
Sundqvist, L., Keenan, K., Zackrisson, M., Prodöhl, P. & Kleinhans, D. Directional genetic differentiation and relative migration. Ecol. Evol. 6, 3461–3475 (2016).
Acknowledgements
This research was supported by a Japan Society for the Promotion of Science Kakenhi Grant-in-aid for Young Scientists (A) 17H04996 to NY, and by the KAIGIN Environment Fund 2014–2015 (managed by the Okinawa Kaiho Bank, Ltd.) and the PRO NATURA FUND 2014 (mannaged by Pro Natura Foundation Japan) to YF.
Author information
Authors and Affiliations
Contributions
A.I. and Y.F. conceived the study. T.Y., N.Y. and A.I. analysed data. T.Y. wrote the first draft of the manuscript, and A.I., N.Y., Y.Y., T.S. and Y.F. authored or reviewed drafts of the paper, approved the final draft. N.Y. and Y.Y. contributed to molecular experiments. T.S. and Y.F. contributed to field survey.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yorisue, T., Iguchi, A., Yasuda, N. et al. Evaluating the effect of overharvesting on genetic diversity and genetic population structure of the coconut crab. Sci Rep 10, 10026 (2020). https://doi.org/10.1038/s41598-020-66712-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-66712-4
- Springer Nature Limited
This article is cited by
-
The domestication and breeding history of Castanea crenata Siebold et Zucc. estimated by direction of gene flow and approximate Bayesian computation
Tree Genetics & Genomes (2023)
-
Using density estimates, sex ratios and size structure to assess the status of a threatened Australian freshwater crayfish (Euastacus armatus) population
Hydrobiologia (2023)
-
Identification of source populations for reintroduction in extinct populations based on genome-wide SNPs and mtDNA sequence: a case study of the endangered subalpine grassland butterfly Aporia hippia (Lepidoptera; Pieridae) in Japan
Journal of Insect Conservation (2022)
-
Genetic diversity and variation of seven Chinese grass shrimp (Palaemonetes sinensis) populations based on the mitochondrial COI gene
BMC Ecology and Evolution (2021)