
Abstract
Duplication of chromosome 22q11.2 (LCR A-D) has been reported at higher frequencies in clinical samples than the general population, but phenotypes vary widely. Triplication (4 copies) is rare, but studying the associated phenotype may provide insight into dosage-sensitivity of the genes in this chromosomal interval. We describe a proband with a triplication, specifically a “double duplication” (two copies per chromosome) of the 22q11.2 region, while his parents and two siblings each have a single duplication (3 copies). The proband had a heart malformation, dysmorphic features, and learning and socialization deficits, whereas the other family members did not. This family illustrates that while duplication of the 22q11.2 may not be sufficient to cause clinically significant neurodevelopmental or health-related phenotypes, triplication of the same region may result in a phenotype characterized by a mild neurodevelopmental disorder, facial dysmorphism, and possibly cardiac anomalies.
Similar content being viewed by others


Introduction
The 22q11.2 chromosomal region is susceptible to rearrangements due to the presence of several low-copy repeat sequences.1 These sequences facilitate non-allelic homologous recombination (NAHR) between chromosomes leading to deletions and duplications, typically 1.5 to 3 megabases (Mb) in length.1 The 22q11.2 deletion syndrome has long been recognized as a heterogeneous condition characterized by congenital heart defect, cleft palate or velopharyngeal insufficiency, developmental delay, and a heightened predisposition to psychiatric disorders.2 The first individual with a duplication of the 22q11.2 region (3 copies) detected by fluorescence in situ hybridization (FISH) was described in 1999.3 Since then, over 50 additional cases have been reported, with highly variable features.4 Early reports were identified by FISH analysis for suspicion of 22q11.2 deletion syndrome (22q11.2 DS), and therefore were biased towards those with similar features.5 Clinical features reported in individuals ascertained by chromosomal microarray (CMA), in contrast, are often mild and highly variable, including cognitive and behavioral issues, congenital malformations, facial dysmorphism, and other health problems.4 The duplication is inherited from a parent in at least 70% of reported cases.4
Here we describe a family of five, ascertained via the proband with atypical autism who carries a triplication (4 copies) of the 22q11.2 region, specifically two copies on each chromosome; herein referred to as “double duplication” (2:2). Both parents and the unaffected siblings carry the 22q11.2 duplication (2:1). To our knowledge, this is the first reported case of 22q11.2 double duplication. We describe in detail the clinical features of the proband, who clinically resembles a previously reported child with a triplication (3:1) at 22q11.2.
Results
Molecular characterization of family
CMA in the proband showed four copies of the 22q11.2 region from nucleotide position 18,890,162 to 21,440,515 (hg19). Genotype analysis confirmed the parental self-reporting of non-relatedness to each other (data not shown). The father has a copy number gain at 22q11.21 of 2.73 Mb from nucleotide position 18,713,432 to 21,440,515 (hg19) and the mother has a copy number gain at 22q11.21 of 2.55 Mb from 18,890,162 to 21,440,515 (hg19). Both siblings of proband had similar gains (Supplementary Figure 1). Copy number variant (CNV) analysis examining sequence read-depth of whole genome sequencing (WGS) data confirmed the duplications (data not shown). FISH testing of both parents confirmed a 2:1 configuration of the copy number gains at 22q11.2 and a 2:2 configuration in the proband (Supplementary Figure 1). Using the WGS data, the allele ratio of informative SNP genotypes within the 22q11 duplication (where both parents are homozygous, but had different genotypes with the proband being heterozygous), confirmed this interpretation (Supplemental information Fig. 2).
Whole genome sequencing of the proband's DNA identified putative loss-of-function variants or compound heterozygous variants in the autism spectrum disorder (ASD) candidate genes MSR1 and SDC2, as well as MYO1A and CLTCL1, respectively. Moreover, a de novo frameshift mutation in SMAD6 (p.Ala424Profs*115) was observed (all mutations are described in Supplemental information Table 1). Using methods described previously6, 7, no additional rare coding CNVs were detected.
Clinical characterization of the family
II-1: Proband (double duplication 22q11.2)
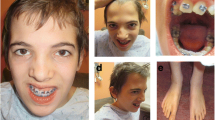
The proband (II-1 in Fig. 1) is a 10-year-old male with a history of exotropia, pulmonary valve stenosis, facial dysmorphism, learning disability, and ASD. He was the first child born to a non-consanguineous Caucasian couple from South Africa. Pregnancy was unremarkable. Delivery was induced at 36.5 weeks’ gestation due to oligohydramnios of unknown cause. Birth weight was 2353 g (10th percentile). An echocardiogram revealed mild pulmonary valve stenosis. Hearing and vision have been normal. At 7 years of age, weight, height, BMI, and head circumference were all at the 25th percentile. Dysmorphic features were present (Fig. 1). Development was notable for mild delay in independent walking (18 months) and more significant delay in expressive speech, with first clear words around 3½ years. He did not demonstrate craniosynostosis as might be suggested by the de novo frameshift mutation found in SMAD6 (Supplemental information Table 1).

Pedigree of family and facial photographs of proband (II-1). The parents provided written informed consent for publication of photographs. Note distinctive facial features including hooded eyelids a, long and smooth philtrum with a thin upper lip a and c, low-set and posteriorly rotated ears b and d with preauricular pits and forward facing earlobes
At age 6 years, he was diagnosed with a learning disability related to processing speed and working memory deficits, challenges with visual memory, and ASD. At 8 years old, a repeat assessment for educational planning showed superior verbal reasoning and average nonverbal reasoning skills and persistent deficits in processing speed and working memory. Psychological assessment results are summarized in Table 1.
I-1: Father (duplication 22q11.2)
The father of the proband (I-1 in Fig. 1) is a 41-year-old man who denies medical problems. He attended regular classes at grade level in both elementary and secondary school. He is a vocational college graduate working full time. On examination, he had a left pre-auricular pit, a long, flat philtrum, and a wide uvula. Details of psychological assessment done at age 38 years are found in Table 1. On tests of intellectual functioning and academic skills, he scored within the average range.
I-2: Mother (Duplication 22q11.2)
The mother of the proband (I-2 in Fig. 1) is a 44-year-old woman who denies medical problems. As a child she was in regular classes at grade level. She is a vocational college graduate who formerly worked as a medical receptionist. On examination, no dysmorphic features were observed. Details of psychological assessment done at age 41 years are found in Table 1. On tests of intellectual functioning and academic skills, she scored within the average range for verbal reasoning and academic skills, and low average for perceptual reasoning skills. Clinically significant symptoms of anxiety and depression were endorsed on a self-report measure.
II-2 and II-3: Sisters (duplication 22q11.2)
The sisters of the proband (II-2 and II-3 in Fig. 1) are monozygotic twins. They have no significant past medical history and developmental milestones were met within expected age norms. They are currently in regular grade 3 functioning academically at grade level. At age 8 years, both have average growth parameters; both have bilateral preauricular ear pits but no other dysmorphic features. Details of psychological assessment at age 8 years are found in Table 1. Both girls have average overall intellectual abilities and academic skills, and both endorsed clinically elevated symptoms of social anxiety.
Discussion
We present the first reported case of “double duplication” of the 22q11.2 region. Probands with this duplication have a wide range of phenotypes affecting physical and cognitive development, and the duplication is frequently inherited from a parent.4 Thus, it remains unclear whether increased dosage of this region truly causes a recognizable phenotype. Some authors have speculated that it could be a benign polymorphism or a risk variant which requires a “second hit” in order to result in a clinically significant phenotype.8 A second hit could, in theory, be an additional copy of the same region, assuming that one or more of the involved genes are dosage sensitive. The triplicated region in our proband contains 66 RefSeq genes, including TBX1 and CRKL; cardiac defects in the 22q11.2 deletion syndrome have been attributed to haploinsufficiency of these two genes.9, 10 Our proband had pulmonic stenosis, while another triplication proband had sub-aortic stenosis and a ventricular septal defect.11, 12 Although experimental evidence is lacking, it is plausible that increased dosage of these genes could also predispose to aberrant cardiac development. The only other report of a proband with a 22q11.2 triplication, also (3:1), is an 8-year-old girl with a strikingly similar phenotype to our proband: learning difficulties, social immaturity, and similar dysmorphic features.13
Another possible reason for our proband’s phenotype, in particular his autistic features, is the contribution of additional mutations in autism candidate genes. We investigated this by analyzing the genome sequence of the proband and his parents using our variant pipeline optimized for identification of ASD-related genes14; however, we are yet to identify any convincing additional mutations that would be clinically characterized as being pathogenic for ASD. Alternatively, it is possible that increased copy number at 22q11.2 could contribute to vulnerability to ASD; at least one study has reported a relatively high prevalence of autism in probands with 22q11.2 duplication.15 From a genomic point of view, this case could be considered as an example of a recessive disorder caused by copy number gains, since the parents carrying three copies are normal, but the proband carrying four copies presents with a phenotype.
In conclusion, we describe in detail the phenotype of a boy with four copies of the 22q11.2 region, and his four first-degree relatives who carry the duplication (three copies) to illustrate the effects of increased copy number of the 22q11.2 region. This family provides support for the view that the 22q11.2 duplication may not be pathogenic on its own and require additional rare and/or common variants, perhaps in unique combinations, to express phenotype.16,17,18,19,20 However, a triplication (either by double duplication, as in our proband, or by expansion of a parental duplication) of the region may result in a phenotype affecting cardiovascular development, facial appearance, cognition, and social communication.
Methods
CMA by array CGH (4 × 180 K ISCA design, Oxford GeneTechnology, Oxford, UK) was performed on all five family members. The proband and both parents also had FISH testing with TUPLE/HIRA1 probe (Vysis, Abbott Park, Illinois, USA). As part of a separate research study, the proband and both parents had WGS as described recently with the data available in the Autism Speaks-supported MSSNG whole genome sequence and phenotype database.14, 21 All sequence data can be accessed through the MSSNG database sponsored by Autism Speaks on Google Genomics (https://www.mss.ng). The proband and family members underwent psychological assessment and examination by a clinical geneticist. The methods were performed in accordance with relevant guidelines and regulations and approved by the Research Ethics Board at the Hospital for Sick Children (Toronto, Canada). The parents provided written informed consent for participation and publication of photographs.
Data availability
The data, as part of a larger autism whole-genome sequencing project, are available in the MSSNG database on Google Genomics (for access see http://www.mss.ng/researchers).
References
McDermid, H. E. & Morrow, B. E. Genomic disorders on 22q11. Am. J. Hum. Genet. 70, 1077–1088 (2002).
Gambini, O. Psychiatric disorders associated with 22q11.2 deletion syndrome. Ment. Illn. 8, 6590 (2016).
Edelmann, L. et al. A common molecular basis for rearrangement disorders on chromosome 22q11. Hum. Mol. Genet. 8, 1157–1167 (1999).
Firth, H. V. 22q11.2 Duplication. in GeneReviews(®) (eds Pagon, R. A. et al.) (University of Washington, 1993).
Ensenauer, R. E. et al. Microduplication 22q11. 2, an emerging syndrome: clinical, cytogenetic, and molecular analysis of thirteen patients. Am. J. Hum. Genet. 73, 1027–1040 (2003).
Pinto, D. et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 94, 677–694 (2014).
Zarrei, M., MacDonald, J. R., Merico, D. & Scherer, S. W. A copy number variation map of the human genome. Nat. Rev. Genet. 16, 172–183 (2015).
Courtens, W., Schramme, I. & Laridon, A. Microduplication 22q11.2: A benign polymorphism or a syndrome with a very large clinical variability and reduced penetrance?—Report of two families. Am. J. Med. Genet. A. 146A, 758–763 (2008).
Racedo, S. E. et al. Mouse and human CRKL is dosage sensitive for cardiac outflow tract formation. Am. J. Hum. Genet. 96, 235–244 (2015).
Racedo, S. E. et al. Reduced dosage of β-catenin provides significant rescue of cardiac outflow tract anomalies in a Tbx1 conditional null mouse model of 22q11.2 deletion syndrome. PLoS. Genet. 13, e1006687 (2017).
Pires, R. et al. Screening of copy number variants in the 22q11.2 region of congenital heart disease patients from the São Miguel Island, Azores, revealed the second patient with a triplication. BMC Genet. 15, 115 (2014).
Vaz, S. O. et al. A unique phenotype in a patient with a rare triplication of the 22q11.2 region and new clinical insights of the 22q11.2 microduplication syndrome: a report of two cases. BMC Pediatr. 15, 95 (2015).
Yobb, T. M. et al. Microduplication and triplication of 22q11.2: a highly variable syndrome. Am. J. Hum. Genet. 76, 865–876 (2005).
Yuen, C., R. K. et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 20, 602–611 (2017).
Wenger, T. L. et al. 22q11.2 duplication syndrome: elevated rate of autism spectrum disorder and need for medical screening. Mol. Autism 7, 27 (2016).
Costain, G. et al. Adult neuropsychiatric expression and familial segregation of 2q13 duplications. Am. J. Med. Genet. Part B 165B, 337–344 (2014).
Peyrot, W. J., Robinson, M. R., Penninx, B. W. J. H. & Wray, N. R. Exploring boundaries for the genetic consequences of assortative mating for psychiatric traits. JAMA Psychiatry 73, 1189–1195 (2016).
Zhu, Z., Lu, X., Yuan, D. & Huang, S. Close genetic relationships between a spousal pair with autism-affected children and high minor allele content in cases in autism-associated SNPs. Genomics 109, 9–15 (2017).
Weiner, D. J. et al. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat. Genet. https://doi.org/10.1038/ng.3863 (2017).
Woodbury-Smith, M. et al. Variable phenotype expression in a family segregating microdeletions of the NRXN1 and MBD5 autism spectrum disorder susceptibility genes. Npj Genomic Med. 2, 17 (2017).
Jiang, Y. et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am. J. Hum. Genet. 93, 249–263 (2013).
Acknowledgements
The authors thank the family for participating in our study and consenting to publish this report. We are grateful to Gurpreet Kamboj and Jessica Suarez for assistance. This study was supported by the Physicians’ Services Incorporated Foundation (PSI 13-43), Genome Canada, the Canadian Institutes of Health Research (CIHR), Autism Speaks, and the University of Toronto McLaughlin Centre. S.W.S. Holds the GlaxoSmithKline-Chair in Genome Sciences at the Hospital for Sick Children and University of Toronto.
Author information
Authors and Affiliations
Contributions
M.T.C. and I.D. conceived and designed the study. M.T.C., I.D., B.M.M., and S.J. performed clinical assessments. All authors contributed to data acquisition, analysis, and interpretation. B.T. and S.W. analyzed the genomic data. B.D. and M.T.C. drafted the manuscript. All authors contributed to critical revision of the manuscript for intellectual content and final approval of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
Cite this article
Dale, B., Modi, B.M., Jilderda, S. et al. Atypical autism in a boy with double duplication of 22q11.2: implications of increasing dosage. npj Genomic Med 2, 28 (2017). https://doi.org/10.1038/s41525-017-0031-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-017-0031-6
- Springer Nature Limited