

Abstract
The incorporation of defects, such as vacancies, into functional materials could substantially tailor their intrinsic properties. Progress in vacancy chemistry has enabled advances in many technological applications, but creating new type of vacancies in existing material system remains a big challenge. We show here that ionized nitrogen plasma can break bonds of iron-carbon-nitrogen-nickel units in nickel-iron Prussian blue analogues, forming unconventional carbon-nitrogen vacancies. We study oxygen evolution reaction on the carbon-nitrogen vacancy-mediated Prussian blue analogues, which exhibit a low overpotential of 283 millivolts at 10 milliamperes per square centimeter in alkali, far exceeding that of original Prussian blue analogues and previously reported oxygen evolution catalysts with vacancies. We ascribe this enhancement to the in-situ generated nickel-iron oxy(hydroxide) active layer during oxygen evolution reaction, where the Fe leaching was significantly suppressed by the unconventional carbon-nitrogen vacancies. This work opens up opportunities for producing vacancy defects in nanomaterials for broad applications.
Similar content being viewed by others

Introduction
Prussian blue analogues (PBAs) are a class of perovskite-type materials described by the general formula \({\mathrm{A}}_{\mathrm{x}}{\mathrm{M}}\left[ {{\mathrm{Fe}}\left( {{\mathrm{CN}}} \right)_6} \right]_{\mathrm{y}} \cdot {\mathrm{mH}}_{\mathrm{2}}{\mathrm{O}}\left( {0 \le {\mathrm{x}} \le 2,{\mathrm{y}} < 1} \right)\), where A is alkali metal and M is transition metal1,2,3. In such materials, nitrogen-coordinated M cations and carbon-coordinated Fe sites are bridged by cyanide (CN) groups, forming open frameworks. Research on PBAs reveals that their intriguing structures enable diverse applications, such as molecule-based magnets4,5, sensors6,7, hydrogen storage8,9 and metal-ion batteries1,10,11,12. In recent years, PBAs have also emerged as catalytic materials for oxygen evolution reaction (OER)13,14,15—a key enabling process for rechargeable metal–air batteries and photo/electrochemical water splitting16,17. For examples, Galan-Mascaros et al.14 described decent OER activity on Co–Fe PBA catalyst, which was later found to be stable in acid through suitable chemical etching treatment14. Zhang et al. used operando X-ray spectroscopy on Ni–Fe PBA to identify the catalytically active sites, suggesting that amorphous Ni hydroxide after OER contributes to the reactivity15. Nevertheless, the role of Fe species in the PBA catalysts for OER is unclear. Moreover, engineering the structure of these CN-bridged bimetallic open frameworks may lead to further enhancements in OER catalysis, which, however, has rarely been researched.
Manipulating defect chemistry can tune the properties and functionalities of materials, such as band structure, conductivity, magnetism, and catalysis18,20,20. The most common defect observed is vacancy, and typically, vacancy engineering can lead to substantial structural perturbations in catalysts, with the capability to tailor surface electronegativity, charge concentration, and redistribution18,19. Once overcoming their formation energy, both anion and/or cation vacancies can be formed, giving diverse vacancy defects like oxygen21,22, sulfur20,23, iodine24, nickel25, iron26, and sometimes dual vacancies27,28. PBAs are the well-known materials bearing Fe(CN)6 vacancies that permit the alteration of electron transfer phenomena1,2. In principle, a new type of vacancy created in PBAs would trigger the change of local electronic environment, which subsequently leads to regions of enhanced energetics for catalysis. But no previous attempts were seen to produce unconventional vacancies in PBA materials, mainly owing to the lack of effective synthetic pathways.
Here, we report the generation of unusual CN vacancies (denoted as VCN) in the Ni–Fe PBA (i.e., K2NiFe(CN)6) through a nitrogen plasma bombardment. Despite the strong affinity of metals and CN ligands, our comprehensive characterizations confirm that the ionized N2 can overcome the energy of VCN formation, yielding unprecedented VCN. The obtained Ni–Fe PBA catalyst with VCN enables highly efficient OER catalysis, far exceeding that of original sample and also previously reported vacancy-mediated OER catalysts. Besides offering tailored local electronic features, our another key finding is that the presence of VCN can suppress the release of Fe species, which therefore favors the growth of Ni–Fe oxy(hydroxide) active layer via a dynamic self-reconstruction of the PBA pre-catalyst during OER. Our work widens the family of vacancy defects, which provides access to enhanced functionalities of PBA materials for various applications.
Results
Synthesis and characterization of VCN-mediated Ni–Fe PBA
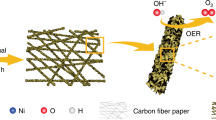
We started with the synthesis of NiMoO4 nanorods acted as precursors using a hydrothermal method described by us previously29,30 (see the Methods section; Supplementary Fig. 1). The NiMoO4 was then dispersed in a K4Fe(CN)6•3H2O aqueous solution under ultrasonication for 2 h, which templates the formation of one-dimensional K2NiFe(CN)6 Prussian blue analogues (PBAs; Supplementary Fig. 1). Consequently, we used N2 plasma bombardment to trigger the VCN formation, as shown schematically in Fig. 1. This process provides energetic nitrogen radicals that enable the cleavage of Fe–C and Ni–N bonds in Fe–CN–Ni units, whereas cleaving the CN triple bond needs to overcome a higher activation barrier; hence the generated CN− diffuses away from the PBA lattices, forming VCN (Fig. 1; Supplementary Fig. 2). Figure 2a shows the transmission electron microscopy (TEM) image of the PBA bombarded with N2 plasma for 60 min (which we abbreviate PBA-60), revealing the rod-like structure with jagged edges. Close-up inspection of these polycrystalline nanorods (inset in Fig. 2a) shows that they actually consist of many interconnected, welded cubes (Supplementary Fig. 3). X-ray diffraction (XRD) analysis uncovers the cubic K2NiFe(CN)6 phase of the pristine PBA (JCPDS 200915; Supplementary Fig. 4). We also bombarded the PBAs with N2 plasma for different times, and do not see noticeable signs of phase, component, or morphological changes compared with the pristine materials (Supplementary Figs 5, 6). These results together indicate that N2 plasma bombardment would not cause structural damage of PBAs, but generates CN vacancies. The Brunauer–Emmett–Teller (BET) surface area of the PBA-60 was determined to be 68.3 m2 g−1, larger than the value of pristine PBAs (i.e., PBA-0) owing to the more accessible vacancy sites (Supplementary Fig. 7).

Schematic illustration of the preparation of VCN-mediated Ni–Fe PBA. Top: VCN forms in Ni–Fe PBA material through N2 plasma bombardment. Bottom: a full diagram of the N2 plasma apparatus

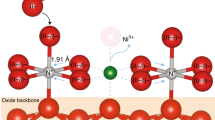
Characterization of VCN-mediated Ni–Fe PBA. a TEM image of PBA-60. Scale bar, 100 nm. Inset shows the corresponding selected-area electron diffraction pattern. Scale bar, 2 nm−1. b HRTEM image of PBA-60. Scale bar, 1 nm. Inset shows the atomic intensity profile along the dotted red line in b. c HAADF image of a typical PBA-60 nanorod. Scale bar, 50 nm. Inset shows the corresponding STEM elemental mappings. Scale bar, 100 nm. d PAS analysis of PBA-0 and PBA-60. e, f Schematic representation of the trapped positrons by VCN-mediated PBA-60
We applied multiple characterization techniques to verify the formation of VCN in PBAs after N2 plasma bombardment. Figure 2b gives a high-resolution TEM (HRTEM) image of PBA-60, which displays crystalline domains with discontinuous atomic arrangement, as further demonstrated by the peak valleys of the atomic intensity profile (inset in Fig. 2b). The local lattice discontinuity suggests the presence of VCN that created by ionized N2 radicals. Scanning TEM (STEM) elemental mapping shows no clear C and N fades throughout the PBA-60 nanorods (Fig. 2c), which reveals that the concentration of VCN does not notably alter the bulk components. We further confirmed the VCN through positron annihilation spectroscopy (PAS), which can offer straightforward structure information of vacancy defects31. In Fig. 2d, the positron lifetime spectra of both PBA-0 and PBA-60 yield three lifetime components, where the two longer components (τ2 and τ3) come from the large voids and the interface in the materials (Supplementary Table 1). The measured shorter component (τ1) of 267 ps for PBA-60 matches well with the calculated positron lifetime of 270 ps for VCN, implying that positron annihilation traps at such vacancies (Supplementary Tables 1, 2). Moreover, our calculation reveals that positrons mainly concentrate at the center of VCN in PBA-60 (Fig. 2e, f).
Considering that vacancy defects are able to tune the photoluminescence (PL) and cathodoluminescence (CL) properties through the mediated band-to-band transition32, we conducted PL and CL measurements on PBA-0 and PBA-60 for comparison. We found clear PL and CL enhancements of PBA-60 versus PBA-0, probably owing to the high quantum efficiency of excitons localized at the VCN sites (Supplementary Figs 8, 9). In addition, we bubbled the ninhydrin and Na2CO3 aqueous solution with tail gas produced during the synthesis process, and observed the fading of original yellow solution, adding support to the formation of CN anions33 (Supplementary Fig. 10). Together, all the above results provide solid evidences that CN vacancies are formed in PBA materials.
OER performance
To explore the influence of new CN vacancies on the OER catalytic behavior, we evaluated the OER activities of PBA-0 and VCN-mediated PBAs in O2-saturated 1 M KOH at ambient environment. The PBAs that bombarded by N2 plasma for 10, 30, 120 min are abbreviated hereafter as PBA-10, PBA-30, and PBA-120. We recorded the OER polarization curves for comparison until the activity becomes stable (Supplementary Fig. 11). Figure 3a shows that all the PBA pre-catalysts exhibit a distinct peak at ~1.38 V originating from the Ni2+/Ni3+ redox process34,35. We found that PBA-0 requires an overpotential of 440 mV at 10 mA cm−2 (normalized on the basis of geometrical surface area), which decreased to 368 mV for PBA-10, 321 mV for PBA-30, 283 mV for PBA-60, and 310 mV for PBA-120 (Fig. 3a). Thus, the formation of VCN in PBA plays a key role for improving the OER energetics. Note that the small overpotential of 283 mV at 10 mA cm−2 for PBA-60 is 32 mV and 50 mV lower than that of the state-of-the-art NiFe-layered double-hydroxide (NiFe–LDH)36 and 20 wt% Ir/C catalysts (Fig. 3a; Supplementary Figs 12, 13), respectively. Tafel plots (Fig. 3b) indicate that PBA-60 has a very small Tafel slope of 54 mV dec−1, lower than 108 and 69 mV dec−1 for PBA-0 and NiFe–LDH catalysts, respectively. Meanwhile, at the overpotential of 300 mV, PBA-60 has a 31-fold improvement in current density versus PBA-0, although the increase in BET surface area by a factor of mere ~1.2 (Supplementary Fig. 7). The predominant OER properties of PBA-60 over other studied catalysts are also clearly seen at overpotentials of 320 mV and 340 mV (Fig. 3c). These results again suggest that VCN should be the main reason that results in the remarkable OER energetics of PBA-60.

OER evaluation. a OER polarization curves of different studied catalysts. Catalyst loading: ~0.255 mg cm−2. Sweep rate: 5 mV s−1. b Tafel plots of different catalysts derived from polarization curves shown in Supplementary Fig. 13. Tafel plot of NiFe–LDH derived from the polarization curve of the cathodic sweep to avoid the interference of the redox peak. c Comparison of the current densities generated on different catalysts at overpotentials of 300 mV, 320 mV, and 340 mV, respectively. The data were extracted from the polarization curves shown in Supplementary Fig. 13. d EIS Nyquist plots of different catalysts at 400 mV overpotential without iR-correction. e Arrhenius plot of the OER kinetic current on different catalysts at 300 mV overpotential without iR-correction. f Comparison of onset potential (defined as the potential at 1 mA cm−2) and overpotential at 10 mA cm−2 for various OER catalysts with vacancy defects. Values were plotted from references where they are reported as such (Supplementary Table 3). All OER measurements were performed in O2-saturated 1 M KOH electrolyte, and the reported data were iR-corrected unless otherwise stated
To probe the charge transfer resistance (Rct) of studied catalysts, we conducted electrochemical impedance spectroscopy (EIS) at 400 mV overpotential, which offers the information regarding the number of electrons transferred from the catalyst surface to the reactant. Figure 3d reveals that the Rct of PBA-60 is 11 Ohms, substantially smaller than that of ~74 Ohms for PBA-0 and ~32 Ohms for the NiFe–LDH catalyst (Supplementary Fig. 13b). This result matches well with our double-layer capacitance (Cdl) measurements that give the highest Cdl value for PBA-60 catalyst (Supplementary Fig. 14). We then studied the influence of temperature on the OER performance of different catalysts at an overpotential of 300 mV to assess their kinetic barriers16. We observed a linear relationship between 293 K and 323 K that follows the Arrhenius behavior, from which the activation energies of 67 kJ mol−1, 36 kJ mol−1, and 44 kJ mol−1 were extracted for PBA-0, PBA-60, and NiFe–LDH catalysts, respectively (Fig. 3e; Supplementary Fig. 15). The lowest apparent barrier value measured for PBA-60 provides evidence that OER kinetics on PBA is highly promoted by CN vacancies. In addition, the effect of plasma treatment atmosphere and power on OER activity showed that PBA-60 obtained by N2 plasma bombardment at a power of 100 W has the optimal OER activity (Supplementary Figs 16–19).
Compared with previously reported catalysts with diverse vacancy defects, we underscore that our PBA-60 shows the best OER performances in terms of the overpotential at 10 mA cm−2 and the onset than were shown before (Fig. 3f; Supplementary Table 3). We believe that the new VCN engineering of other materials can guarantee predictable better-performing OER catalysts.
Chemical and structure alterations
We now study the component evolution of PBA with time during the N2 plasma bombardment. We applied the inductively coupled plasma atomic emission spectroscopy (ICP-AES; measuring K, Fe, and Ni) and elemental analysis (measuring C and N) to probe the bulk element variation, whereas the surface component evolution was determined by the X-ray photoelectron spectroscopy (XPS). We found that both bulk and surface atomic ratios of K/Ni, Fe/Ni, and C/N remain unchanged even bombarding for 60 min, but the atomic ratios of N/Ni and C/Fe decreased with time (Supplementary Fig. 20, Supplementary Tables 4, 5). This unambiguously suggests the formation of sole VCN in PBA without any other vacancy defects. However, when we bombarded the PBA for 120 min, we observed a decreased Fe/Ni atomic ratio, indicative of some Fe vacancies formed in the structure (Supplementary Fig. 20). Because no Ni loss during N2 plasma process, we are able to calculate the VCN content of different samples based on the measured N/Ni atomic ratios. The bulk VCN contents of ~3.2, 4.5, 7.5, and 11.4% are determined for PBA-10, PBA-30, PBA-60, and PBA-120, slightly lower than that of surface VCN contents (Fig. 4a). Moreover, our Ni and Fe K-edge extended X-ray absorption fine structure (EXAFS) fittings reveal that, after bombarding with N2 plasma for 60 min, the first-shell Ni–N coordination number decreases from 5.9 to 5.5 and the Fe–C coordination number decreases from 6.1 to 5.7 (Fig. 4b; Supplementary Figs 21–24, Supplementary Table 6). Such unsaturated local atomic environment that comes from the formation of VCN could promote the energetics of the OER18.

Chemical and structure alterations of the VCN-mediated PBA catalysts. a The bulk and surface VCN contents of different VCN-mediated PBA catalysts. b Comparison of Ni–N and Fe–C coordination numbers for PBA-0 and PBA-60 catalysts. c Raman spectra of PBA-0 and PBA-60 catalysts. d, e Ni L3-edge and Fe L3-edge XAS spectra of PBA-0 and different VCN-mediated PBA catalysts, respectively. f ESR spectra of PBA-0 and different VCN-mediated PBA catalysts
To further understand the source of high OER activity, we monitored the oxidation states and electronic structure of PBA catalysts using Raman spectroscopy, X-ray absorption spectroscopy (XAS), and XPS. Our Raman spectroscopy measurement on PBA-0 (Fig. 4c) exhibits two prominent peaks at 2098 cm−1 and 2134 cm−1, corresponding to the vibrations of CN groups in the mixture of FeII–CN–NiII and FeII–CN–NiIII, whereas the peak at 2225 cm−1 comes from the CN vibration in FeIII–CN–NiII, in agreement with previous reports10,11. We found that the peak at 2225 cm−1 disappears for PBA-60 sample, meaning the changed oxidation state of FeIII species. Because XAS technique is sensitive to the local structure and chemical environment, we performed Ni L-edge XAS measurements on different PBA samples with NiO and LiNiO2 as references (Supplementary Fig. 25). As Fig. 4d shows, the Ni L3-edge shoulder peak shifts from 855.1 eV (PBA-0) to 855.6 eV (PBA-60), indicating that surface Ni2+ ions were partially oxidized to Ni3+ after N2 plasma bombardment37,38 (Supplementary Fig. 25). The Fe L3-edge XAS spectra in Fig. 4e and Supplementary Fig. 26 reveal two peaks that originated from electron transitions to unoccupied eg orbitals and the additional transitions to anti-bonding π* states39 (inset in Fig. 4e). The increased intensity of the left peak demonstrates lowered oxidation state of Fe with the formation of VCN. The above oxidation-state analyses match well with our XPS studies presented in Supplementary Fig. 27. Further, electron spin resonance (ESR) measurements show that an ESR signal appears at g-factor = 2.03 just after 10 min of bombardment, which becomes dominant when bombarding for 60 min (Fig. 4f). This symmetrical ESR signal originates from the unpaired electrons of Ni3+ (t2g6eg1) species25,40, again indicating the increase in Ni oxidation state after incorporating the CN vacancies.
We now come to an understanding that the new VCN sites not only cause local unsaturated coordination environment but also modulate the oxidation states of Ni and Fe of PBAs. We next use the accumulated knowledge on Ni–Fe oxide OER catalysts to propose potential OER enhancement mechanism of our Ni–Fe PBAs, considering the similarity in their real active sites. Recent studies have demonstrated that Fe sites in Ni–Fe oxides are active, while Ni species act as electrically conductive and chemically stable host for the Fe sites41,42. In our fresh PBA-0 catalyst (i.e., K2NiFe(CN)6), both Ni and Fe are in the oxidation state of 2 + (Fig. 4d; Supplementary Fig. 28)11. The partial metastable Fe2+ is readily oxidized to Fe3+ in the lab environment11,43, which returns to its original state after the VCN formed (Fig. 4c, e). However, the oxidation state of Ni in PBAs was partially boosted to 3 + through VCN mediation (Fig. 4d, f), which suggests the electron transfer from Ni to the adjacent Fe sites. During OER, the VCN-mediated Ni–Fe PBA catalyst with modified metal oxidation state thus energetically transfers to NiFeOOH active layer via surface self-reconstruction (discuss later), giving rise to the marked OER activity.
Our experiments reveal that the OER activity of VCN-mediated Ni–Fe PBA catalyst improves monotonously until the bombarding time reaches 60 min, where it gains a bulk VCN content of ~7.5% (Figs 3a, 4a). Further extending the bombarding time, however, causes the loss of Fe in the structure (Supplementary Fig. 20 and Tables 4, 5), leading to inferior OER properties. We further prepared Co–Fe PBA nanorods using the same methodology and created VCN through N2 plasma bombardment (Supplementary Fig. 29). We also observed the VCN-mediated OER improvement on Co–Fe PBAs, but its activity is largely lower than that of Ni–Fe PBAs (Supplementary Fig. 30), which demonstrates that Ni–Fe-based OER catalysts are notable, agreeing with previous reports44.
Fe leaching suppression by the VCN
Figure 5 illustrates another critical observation that we want to demonstrate in this work: that is, the Fe loss in Ni–Fe PBA catalysts is substantially suppressed in the presence of CN vacancies. Researchers have previously seen that [Fe(CN)6]4− groups in PBAs readily diffuse outward to the electrolytes during OER, causing Fe loss in the structure14,15. Our operational stability assessments exhibit that PBA-60 catalyst performs very robustly, even at a high current density of 1 A cm−2, whereas the PBA-0 decays gradually with time (Fig. 5a; Supplementary Fig. 31). Energy-dispersive X-ray spectroscopy (EDX) analyses reveal that Fe suffers a certain loss at the first 5 h of cycling for PBA-60, which is retained thereafter even up to 25 h (Fig. 5b; see Supplementary Fig. 32 for the detailed Fe/Ni atomic ratios). In contrast, the PBA-0 loses most Fe in the structure after 25 h of continuous operation. These results agree well with our STEM elemental mapping studies shown in Fig. 5d–g. Moreover, the time-dependent Fourier transform infrared (FT-IR) spectra on PBA-60 catalyst exhibit that three bands at 3650 cm−1, 880 cm−1, and 640 cm−1 become dominant with time (Fig. 5c; Supplementary Fig. 33), which belongs to the O–H stretching mode in brucite-like Ni(OH)2 structure45, as well as the Fe–O–H46 and Ni–O–H45 bending modes, respectively, indicating the formation of Ni–Fe oxy(hydroxide) active surface layer during OER. Note that the Fe–O–H bond at 880 cm−1 was not detected for PBA-0 cycled for 25 h because of the loss of Fe, consistent with above results. The surface self-reconstruction process on our Ni–Fe PBA OER catalyst can be further confirmed by the Raman spectroscopy measurements (Supplementary Fig. 34) and the O K-edge XAS data (Supplementary Fig. 35).

Performance stability and Fe-leaching suppression. a Chronopotentiometric responses recorded on PBA-0 and PBA-60 at a constant current density of 10 mA cm−2. b EDX spectra of PBA-0 and PBA-60 catalysts that cycled for different times. The Cu signals come from Cu-based TEM grid. c FT-IR spectra of PBA-0 and PBA-60 catalysts that cycled for different times. d, e EDX elemental mappings of PBA-0 and PBA-60 catalysts that cycled for 5 h, respectively. Scale bars, 250 nm. f, g EDX elemental mappings of PBA-0 and PBA-60 catalysts that cycled for 25 h, respectively. Scale bars, 100 nm. h Illustrations of the surface reconstruction on the Ni–Fe PBA catalyst during OER, where the VCN enables the suppression of Fe loss, permitting the formation NiFeOOH surface active layer
On the basis of above investigations, we become clear about the origin of the outstanding OER properties on VCN-mediated PBA catalysts. In the Ni–Fe PBA structure, Fe atoms are bonded to six C atoms. The removal of CN groups in PBAs makes the Fe–C coordination numbers decreased, leaving a number of coordinatively unsaturated Fe sites (Fig. 5h). These open metal centers are favorable to bond with oxygen to yield Fe–O bonds during OER, which therefore hampers the Fe loss into the electrolyte. Nevertheless, the fresh PBA-0 catalysts are subjected to a complete anion exchange between [Fe(CN)6]4− and OH− in alkaline solution, which causes significant Fe leaching (Fig. 5h)14,15. Therefore, VCN can largely prevent the Fe loss from PBA catalysts, which leads to the formation of OER-active NiFeOOH surface layer and enhances the OER activity.
Discussion
In conclusion, we have demonstrated that unconventional CN vacancies were generated in Ni–Fe PBA materials via ionized N2 bombardment, as evidenced by multiple characterization techniques. Such VCN is largely distinct from the well-studied vacancies previously documented, which not only tunes the local electronic structure and coordination environment of the Ni–Fe sites, but also limits the loss of Fe element during OER process; these together enable a new, robust, and high-performance Ni–Fe oxyhydroxide from PBA pre-catalyst during OER. We anticipate that this unusual VCN could also be achievable in other PBAs, which thus opens up the possibilities for exploring new applications of PBAs beyond catalysis.
Methods
Material synthesis
The NiMoO4 nanorods were prepared by a hydrothermal method in a pure water system according to our previous works29,30. Briefly, 2 mmol Ni(NO3)2•6H2O, 2 mmol Na2MoO4•2H2O, and 35 mL of H2O were mixed to form a clear solution. The mixture was transferred into a Teflon-lined stainless autoclave (50 mL) and heated at 150 °C for 6 h. After reaction, NiMoO4 powder was obtained by centrifugation, which was then washed and dried for use. Next, 40 mg of NiMoO4 powder was dispersed in the 20 mL K4Fe(CN)6•3H2O (0.3 mmol) aqueous solution with ultrasonication. After 2 h of reaction with drastic stirring at room temperature, the product was centrifuged, washed, and dried to obtain PBA-0. The PBA-0 powder was treated by N2 plasma at various irradiation times of 10 min, 30 min, 60 min, and 120 min with the plasma power of 100 W and the pressure of 3 mTorr. The synthesis of Co–Fe PBAs are the same with the synthesis of Ni–Fe PBAs, except for replacing the Ni(NO3)2•6H2O with Co(NO3)2•6H2O in the first step. The Co–Fe PBA samples were bombarded by N2 plasma for different times, giving desired samples for comparison.
Material characterizations
Morphology of the samples was investigated by TEM (Hitachi H7650) and HRTEM (JEM-ARM 200 F). XRD pattern was obtained from a Philips X’Pert Pro Super X-ray diffractometer equipped using Cu Kα radiation (λ, 1.54184 Å). Raman spectra were carried out at a Raman microscope with a 514 -nm excitation laser. Fourier transform infrared (FT-IR) spectra were recorded on a Fourier transform infrared spectrometer (Bruker Vector 22) with KBr disk method. Photoluminescence (PL) spectra were obtained from a Hitachi F-4600 fluorescence spectrophotometer with an excitation light of 300 nm. Cathodoluminescence (CL) images were obtained from SEM (Sirion200) with equipped with CL system under 10 keV. XPS was taken on an X-ray photoelectron spectrometer (ESCALab MKII) with an X-ray source (Mg Kα hυ = 1253.6 eV). ICP data were obtained by an Optima 7300 DV instrument. N2 adsorption/desorption analysis was conducted on an ASAP 2020 (Micromeritics, USA) at 77 K. ESR spectra were obtained from a JEOL JES-FA200 EPR spectrometer (9065.8 MHz, X band, 300 K). PAS were conducted with a fast-slow coincidence ORTEC system with a time resolution of about 230 ps full width at half-maximum, and our samples were pressed into about 1 -mm-thick disk with a 5 mCi source of 22Na sandwiched between two identical sample disks. The X-ray absorption spectra of Ni and Fe K-edges were obtained from the beamline 1W1B station of Beijing Synchrotron Radiation Facility (China). The X-ray absorption spectra of Ni L-edges and O K-edges were obtained from the 4B9B beamline of Beijing Synchrotron Radiation Facility (China). The X-ray absorption spectra of Fe L-edges were performed on the BL10B beamline of National Synchrotron Radiation Laboratory in Hefei (China).
Electrochemical measurements
The standard three-electrode system was applied to perform the OER electrochemical measurements on the IM6ex electrochemical workstation (ZAHNER elektrik, Germany). A rotating disk electrode (RDE) made of glassy carbon (PINE, 5 -mm diameter, disk area: 0.196 cm2) was used as the working electrode. Platinum foil was used as the counter electrode, and the Ag/AgCl (3.5 M KCl) was used as the reference electrode. The potential difference between Ag/AgCl and RHE was calibrated by the cyclic voltammetry test in H2-saturated 1 M KOH electrolyte (ERHE = EAg/AgCl + 1.03 V).
To make the working electrodes, 5 mg of catalyst powder was dispersed in 960 μL of ethanol with 40 μL of Nafion solution (5 wt%), which was then ultrasonicated to yield catalyst ink. A uniform catalyst film was obtained by pipetting 10 μL of catalyst ink onto the GC electrode, leading to the catalyst loading of ~0.255 mg cm−2. Before OER measurements, the electrolyte (1 M KOH) was bubbled with O2 gas for at least 30 min. The electrodes were pre-cycled between 0 and 0.8 V vs. Ag/AgCl at a sweep rate of 100 mV s−1 for 20 cycles until reaching the stable state, then the OER polarization curves were recorded at a sweep rate of 5 mV s−1 (Supplementary Fig. 11). The EIS measurement was performed over a frequency ranging from 100 KHz to 100 mHz at the amplitude voltage of 5 mV. For the long-term stability test, the catalyst was supported on the carbon fiber paper (1 mg cm−2), which was directly used as the working electrode. All the polarization curves were corrected with iR-correction unless otherwise stated. The onset potential is defined as the potential that yields a current density of 1 mA cm−2.
Data availability
The data that support the findings of this study are available from the corresponding authors upon request.
References
Lee, H.-W. et al. Manganese hexacyanomanganate open framework as a high-capacity positive electrode material for sodium-ion batteries. Nat. Commun. 5, 5280 (2014).
Ma, F., Li, Q., Wang, T., Zhang, H. & Wu, G. Energy storage materials derived from Prussian blue analogues. Sci. Bull. 62, 358–368 (2017).
Aguila, D., Prado, Y., Koumousi, E. S., Mathoniere, C. & Clerac, R. Switchable Fe/Co Prussian blue networks and molecular analogues. Chem. Soc. Rev. 45, 203–224 (2016).
Holmes, S. M. & Girolami, G. S. Sol-gel synthesis of KV[Cr(CN)6] 2H2O: a crystalline molecule-based magnet with a magnetic ordering temperature above 100° C. J. Am. Chem. Soc. 121, 5593–5594 (1999).
Sato, O., Iyoda, T., Fujishima, A. & Hashimoto, K. Photoinduced magnetization of a cobalt-iron cyanide. Science 272, 704–705 (1996).
Karyakin, A. A. et al. Prussian blue based nanoelectrode arrays for H2O2 detection. Anal. Chem. 76, 474–478 (2004).
Lin, Y. et al. A facile electrochemical method for simultaneous and on-line measurements of glucose and lactate in brain microdialysate with Prussian Blue as the electrocatalyst for reduction of hydrogen peroxide. Anal. Chem. 79, 9577–9583 (2007).
Kaye, S. S. & Long, J. R. Hydrogen storage in the dehydrated prussian blue analogues M3[Co(CN)6]2 (M = Mn, Fe, Co, Ni, Cu, Zn). J. Am. Chem. Soc. 127, 6506–6507 (2005).
Krap, C. P., Balmaseda, J., del Castillo, L. F., Zamora, B. & Reguera, E. Hydrogen storage in Prussian Blue analogues: H2 interaction with the metal found at the cavity surface. Energy Fuels 24, 581–589 (2010).
Wang, L. et al. A superior low-cost cathode for a Na-ion battery. Angew. Chem. Int. Ed. 52, 1964–1967 (2013).
Zhang, W. et al. Synthesis of monocrystalline nanoframes of Prussian Blue analogues by controlled preferential etching. Angew. Chem. Int. Ed. 55, 8228–8234 (2016).
Ren, W. et al. Activation of sodium storage sites in Prussian Blue analogues via surface etching. Nano Lett. 17, 4713–4718 (2017).
Pintado, S., Goberna-Ferrón, S., Escudero-Adán, E. C. & Galán-Mascarós, J. R. Fast and persistent electrocatalytic water oxidation by Co–Fe Prussian Blue coordination polymers. J. Am. Chem. Soc. 135, 13270–13273 (2013).
Han, L. et al. Enhanced activity and acid pH stability of Prussian Blue-type oxygen evolution electrocatalysts processed by chemical etching. J. Am. Chem. Soc. 138, 16037–16045 (2016).
Su, X. et al. Operando spectroscopic identification of active sites in NiFe Prussian Blue analogues as electrocatalysts: activation of oxygen atoms for oxygen evolution reaction. J. Am. Chem. Soc. 140, 11286–11292 (2018).
Zhang, B. et al. Homogeneously dispersed, multimetal oxygen-evolving catalysts. Science 352, 333–337 (2016).
Smith, R. D. L. et al. Photochemical route for accessing amorphous metal oxide materials for water oxidation catalysis. Science 340, 60–63 (2013).
Yan, D. et al. Defect chemistry of nonprecious-metal electrocatalysts for oxygen reactions. Adv. Mater. 29, 1606459 (2017).
Schweke, D., Mordehovitz, Y., Halabi, M., Shelly, L. & Hayun, S. Defect chemistry of oxides for energy applications. Adv. Mater. 30, 1706300 (2018).
Li, H. et al. Activating and optimizing MoS2 basal planes for hydrogen evolution through the formation of strained sulphur vacancies. Nat. Mater. 15, 48–53 (2016).
Fabbri, E. et al. Dynamic surface self-reconstruction is the key of highly active perovskite nano-electrocatalysts for water splitting. Nat. Mater. 16, 925–931 (2017).
Kim, H.-S. et al. Oxygen vacancies enhance pseudocapacitive charge storage properties of MoO3-x. Nat. Mater. 16, 454–460 (2017).
Luo, Z. et al. Chemically activating MoS2 via spontaneous atomic palladium interfacial doping towards efficient hydrogen evolution. Nat. Commun. 9, 2120 (2018).
Zhu, X., Lee, J. & Lu, W. D. Iodine vacancy redistribution in organic–inorganic halide perovskite films and resistive switching effects. Adv. Mater. 29, 1700527 (2017).
Zhao, Y. et al. Ultrafine NiO nanosheets stabilized by TiO2 from monolayer NiTi-LDH precursors: an active water oxidation electrocatalyst. J. Am. Chem. Soc. 138, 6517–6524 (2016).
Bin, L. et al. Iron vacancies induced bifunctionality in ultrathin feroxyhyte nanosheets for overall water splitting. Adv. Mater. 30, 1803144 (2018).
Wang, Y. et al. Layered double hydroxide nanosheets with multiple vacancies obtained by dry exfoliation as highly efficient oxygen evolution electrocatalysts. Angew. Chem. Int. Ed. 56, 5867–5871 (2017).
Liu, R., Wang, Y., Liu, D., Zou, Y. & Wang, S. Water-plasma-enabled exfoliation of ultrathin layered double hydroxide nanosheets with multivacancies for water oxidation. Adv. Mater. 29, 1701546 (2017).
Yu, Z.-Y. et al. A one-dimensional porous carbon-supported Ni/Mo2C dual catalyst for efficient water splitting. Chem. Sci. 8, 968–973 (2017).
Yu, Z.-Y. et al. Ni-Mo-O nanorod-derived composite catalysts for efficient alkaline water-to-hydrogen conversion via urea electrolysis. Energy Environ. Sci. 11, 1890–1897 (2018).
Liu, X., Zhou, K., Wang, L., Wang, B. & Li, Y. Oxygen vacancy clusters promoting reducibility and activity of ceria nanorods. J. Am. Chem. Soc. 131, 3140–3141 (2009).
Wu, X., Siu, G., Fu, C. & Ong, H. Photoluminescence and cathodoluminescence studies of stoichiometric and oxygen-deficient ZnO films. Appl. Phys. Lett. 78, 2285–2287 (2001).
Nagaraja, P., Kumar, M. S. H., Yathirajan, H. S. & Prakash, J. S. Novel sensitive spectrophotometric method for the trace determination of cyanide in industrial effluent. Anal. Sci. 18, 1027–1030 (2002).
Trotochaud, L., Young, S. L., Ranney, J. K. & Boettcher, S. W. Nickel–iron oxyhydroxide oxygen-evolution electrocatalysts: the role of intentional and incidental iron incorporation. J. Am. Chem. Soc. 136, 6744–6753 (2014).
Stevens, M. B., Trang, C. D. M., Enman, L. J., Deng, J. & Boettcher, S. W. Reactive Fe-sites in Ni/Fe (oxy)hydroxide are responsible for exceptional oxygen electrocatalysis activity. J. Am. Chem. Soc. 139, 11361–11364 (2017).
Gong, M. et al. An advanced Ni-Fe layered double hydroxide electrocatalyst for water oxidation. J. Am. Chem. Soc. 135, 8452–8455 (2013).
Kang, J.-S. et al. Valence and spin states in delafossite AgNiO2 and the frustrated Jahn-Teller system ANiO2 (A = Li, Na). Phys. Rev. B 76, 195122 (2007).
Zheng, X. et al. Theory-driven design of high-valence metal sites for water oxidation confirmed using in situ soft X-ray absorption. Nat. Chem. 10, 149–154 (2018).
Hocking, R. K. et al. Fe L-edge XAS studies of K4[Fe(CN)6] and K3[Fe(CN)6]: a direct probe of back-bonding. J. Am. Chem. Soc. 128, 10442–10451 (2006).
Davidson, A., Tempere, J., Che, M., Roulet, H. & Dufour, G. Spectroscopic studies of nickel (II) and nickel (III) species generated upon thermal treatments of nickel/ceria-supported materials. J. Phys. Chem. 100, 4919–4929 (1996).
Song, F. et al. Transition metal oxides as electrocatalysts for the oxygen evolution reaction in alkaline solutions: an application-inspired renaissance. J. Am. Chem. Soc. 140, 7748–7759 (2018).
Hunter, B., Winkler, J. & Gray, H. Iron is the active site in nickel/iron water oxidation electrocatalysts. Molecules 23, 903 (2018).
Kulesza, P. J. Solid-state electrochemistry of iron hexacyanoferrate (Prussian Blue type) powders: evidence for redox transitions in mixed-valence ionically conducting microstructures. J. Electroanal. Chem. 289, 103–116 (1990).
McCrory, C. C. L., Jung, S., Peters, J. C. & Jaramillo, T. F. Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J. Am. Chem. Soc. 135, 16977–16987 (2013).
Tian, X. et al. Microwave-assisted non-aqueous homogenous precipitation of nanoball-like mesoporous α-Ni(OH)2 as a precursor for NiOx and its application as a pseudocapacitor. J. Mater. Chem. 22, 8029–8035 (2012).
Zhang, Y., Yang, M., Dou, X.-M., He, H. & Wang, D.-S. Arsenate adsorption on an Fe-Ce bimetal oxide adsorbent: role of surface properties. Environ. Sci. Technol. 39, 7246–7253 (2005).
Acknowledgements
This work was supported by the funding support from the National Natural Science Foundation of China (Grants 21521001, 21431006, 21225315, 21321002, 91645202, and 51702312), the Users with Excellence and Scientific Research Grant of Hefei Science Center of CAS (2015HSCUE007), the Key Research Program of Frontier Sciences, CAS (Grant QYZDJ‐SSW‐SLH036), the Chinese Academy of Sciences (Grants KGZD‐EW‐T05, XDA090301001), the Fundamental Research Funds for the Central Universities (WK2060190045, WK2340000076), and the Recruitment Program of Global Youth Experts. Z.-Y.Y. acknowledges the China Postdoctoral Science Foundation (2018M632545). This work was partially carried out at the USTC Center for Micro and Nanoscale Research and Fabrication.
Author information
Authors and Affiliations
Contributions
M.-R.G., Z.-Y.Y. and S.-H.Y. conceived the idea. S.-H.Y. and M.-R.G. supervised the project. Z.-Y.Y., Y.D. and Y.C. performed the experiments, collected and analyzed the data. T.M. and Y.L. provided the help to analyze the results. J.-D.L. and B.-J.Y. collected and analyzed the PAS data. X.-K.L., W.L. and T.Y. collected and analyzed the XANES data. X.-S.Z. and J.-F.Z. collected and analyzed the XAS data. Z.-Y.Y., M.-R.G. and S.-H.Y. co-wrote the paper. All authors discussed the results and commented on the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information: Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, ZY., Duan, Y., Liu, JD. et al. Unconventional CN vacancies suppress iron-leaching in Prussian blue analogue pre-catalyst for boosted oxygen evolution catalysis. Nat Commun 10, 2799 (2019). https://doi.org/10.1038/s41467-019-10698-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-10698-9
- Springer Nature Limited
We’re sorry, something doesn't seem to be working properly.
Please try refreshing the page. If that doesn't work, please contact support so we can address the problem.